Heterogeneous catalysis
Kinetic Monte Carlo simulations applied to heterogeneous processes of interest in industry applications.
Description
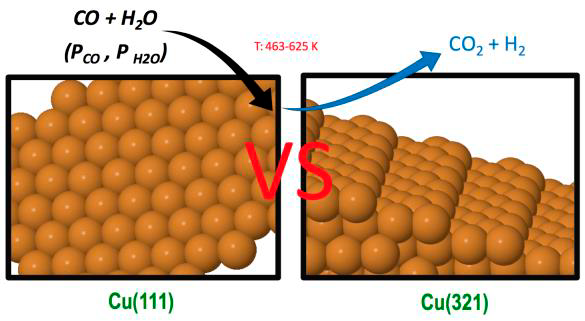
Low coordinated sites play an important role in many heterogeneously catalysed reactions. Step sites are often crucial since, for instance, they allow different types of binding to the catalyst surface, and facilitate bond breaking of reactants. It is generally assumed that the presence of steps enhances the catalytic activity towards a complex reaction by increasing the reactant adsorption energies and decreasing the energy barriers. However, even if the energy barriers for the rate-determining steps (RDS) at step sites are lower than for regular sites, it is not always possible to conclude that the presence of these sites is beneficial. In fact, a strong chemisorption of reactants, products or impurities may cause surface poisoning, and may also result in the lowering of the energy barriers for RDSs also in the reverse direction, increasing reverse reaction rates over the forward ones with a concomitant decrease in the catalytic activity. An important catalysed reaction with special technological relevance is the water gas shift reaction (WGSR) that transforms CO and water into carbon dioxide and molecular hydrogen. It is an equilibrium-limited reaction, where CO conversion is favoured at low temperatures due to its exergonicity. As the temperature increases, the equilibrium constant and the final conversion inherently decrease. Hence, this process is usually carried out in two stages: a first one at quite high temperature (300–450°C) favouring fast CO consumption, and a second one at a lower temperature (200–300°C) to reach higher conversions, which are limited by the thermodynamic equilibrium. Copper based catalysts with inclusion of Zn, Cr and Al oxides and CuO/ZnO/Aluminum oxide are used in low temperature reactors, although other metals and supports have also been proposed.
The molecular mechanism for the low temperature catalyzed WGSR involves a rather large number of elementary steps defining the two possible main routes, usually referred to as surface redox and associative COOH mechanisms. Both start from water dissociation; however, in the former carbon dioxide is produced by direct reaction between adsorbed CO and O, whereas in the latter it is generated through a COOH intermediate.
In our group of research we have recently studied the role of step sites in the WGSR catalysed by Cu surfaces using Density Functional Theory (DFT) calculations including the contribution of dispersion terms. Thus, we compared the energy barriers for all the different pathways involved in the two molecular routes for both the stepped Cu(321) surface and the flat Cu(111) one. Although the presence of step sites increases the water adsorption energy and decreases the energy barriers of different processes one cannot rigorously claim that the catalytic activity of the stepped surface is going to be superior to that of the flat Cu(111) surface, where the COOH formation is faster and the water formation has an energy barrier much higher than on the stepped Cu(321) surface. In fact, some previous studies, for instance, for the reverse WGSR and for the WGSR on Cu(hkl) surfaces conclude that catalytic activity follows the order Cu(110) > Cu(100) > Cu(111). However, these claims are based on comparing only energy barriers of some assumed RDSs. Within a microkinetic model, a rather rigorous determination of a RDS can be done through the evaluation of the degree of rate control for the proposed step as defined by Campbell, or by doing a most sophisticated reaction-route graph analysis based on an equivalent electrical circuit. Other similar methods are also available. We performed kinetic Monte Carlo simulations of the WGSR over the Cu(321) surface, based on earlier DFT calculations, to ascertain its catalytic activity in comparison with a previous kMC study over the flat Cu(111) surface. We provide compelling evidence of the usefulness of this important concept in kMC simulations and its general validity beyond microkinetic models.
Collaborations
Prof. Francesc Illas (Dept. Materials Science and Physical Chemistry, UB, Barcelona, Spain)
Fundings
MINECO (Project: CTQ2014-53987-R)
