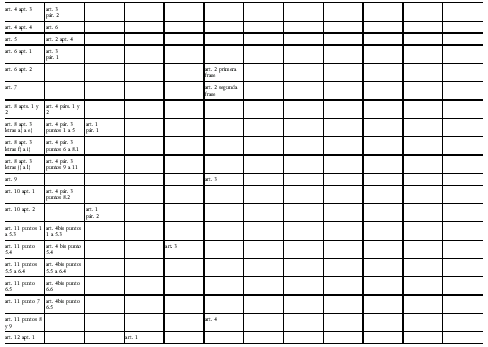
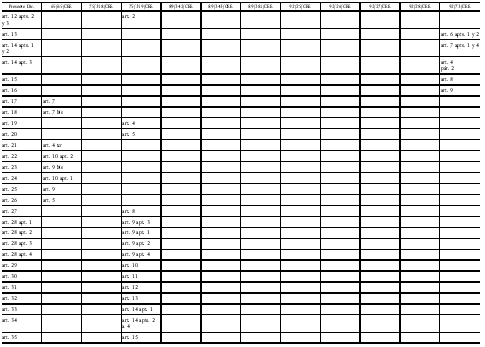
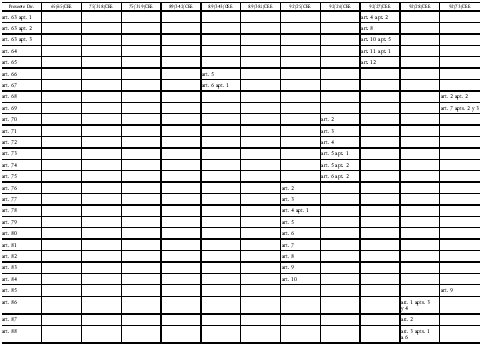
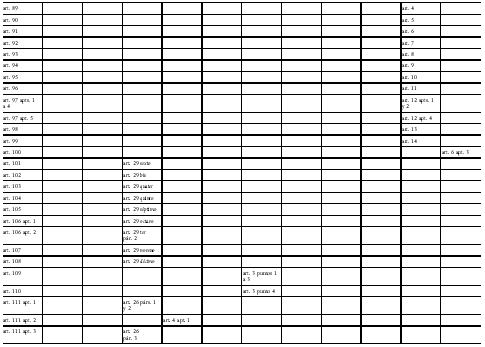
Directiva 2001/83/CE del Parlamento Europeo y del Consejo
de 6 de noviembre de 2001 por la que se establece un código comunitario
sobre medicamentos para uso humano
Modificada por la Directiva 2002/98/CE del Parlamento Europeo y del Consejo, de 27 de enero de 2003, DOCE del 8 de febrero, por la que se establecen normas de calidad y de seguridad para la extracción, verificación, tratamiento, almacenamiento y distribución de sangre humana y sus componentes y por la que se modifica la Directiva 2001/83/CE, y por la Directiva 2003/63/CE de la Comisión, de 25 de junio de 2003, DOCE del 27, que modifica la Directiva 2001/83/CE del Parlamento Europeo y del Consejo por la que se establece un código comunitario sobre medicamentos para uso humano
PARLAMENTO EUROPEO Y EL CONSEJO DE LA UNIÓN EUROPEA,
Visto el Tratado constitutivo de la Comunidad Europea, y en particular su artículo
95,
Vista la propuesta de la Comisión,
Visto el dictamen del Comité Económico y Social(1),
De conformidad con el procedimiento establecido en el artículo 251 del
Tratado(2),
Considerando lo siguiente:
(1) La Directiva 65/65/CEE del Consejo, de 26 de enero de 1965, relativa a
la aproximación de disposiciones legales, reglamentarias y administrativas,
sobre medicamentos(3), la Directiva 75/318/CEE del Consejo, de 20 de mayo de
1975, relativa a la aproximación de las legislaciones de los Estados
miembros sobre normas y protocolos analíticos, tóxico-farmacológicos
en materia de pruebas de medicamentos(4), la segunda Directiva 75/319/CEE del
Consejo, de 20 de mayo de 1975, relativa a la aproximación de disposiciones
legales, reglamentarias y administrativas sobre especialidades farmacéuticas(5),
la Directiva 89/342/CEE del Consejo, de 3 de mayo de 1989, por la que amplía
el ámbito de aplicación de las Directivas 65/65/CEE y 75/319/CEE
y por la que se adoptan disposiciones complementarias sobre medicamentos inmunológicos
consistentes en vacunas, toxinas, sueros y alergenos(6), la Directiva 89/343/CEE
del Consejo, de 3 de mayo de 1989, por la que se amplía el ámbito
de aplicación de las Directivas 65/65/CEE y 75/319/CEE y por la que se
adoptan disposiciones complementarias sobre radiofármacos (89/343/CEE)(7),
la Directiva 89/381/CEE del Consejo, de 14 de junio de 1989, por la que se amplía
el ámbito de aplicación de las Directivas 65/65/CEE y 75/319/CEE
relativas a la aproximación de las disposiciones legales, reglamentarias
y administrativas sobre especialidades farmacéuticas y por la que se
adoptan disposiciones especiales sobre los medicamentos derivados de la sangre
y del plasma humanos(8), la Directiva 92/25/CEE del Consejo, de 31 de marzo
de 1992, relativa a la distribución al por mayor de los medicamentos
para uso humano(9), la Directiva 92/26/CEE del Consejo, de 31 de marzo de 1992,
relativa a la clasificación para su dispensación de los medicamentos
de uso humano(10), la Directiva 92/27/CEE del Consejo, de 31 de marzo de 1992,
relativa al etiquetado y al prospecto de los medicamentos de uso humano(11),
la Directiva 92/28/CEE del Consejo, de 31 de marzo de 1992, relativa a la publicidad
de los medicamentos para uso humano(12), y la Directiva 92/73/CEE del Consejo,
de 22 de septiembre de 1992, por la que se amplía el ámbito de
aplicación de las Directivas 65/65/CEE y 75/319/CEE relativas a la aproximación
de las disposiciones legales, reglamentarias y administrativas sobre medicamentos
y por la que se adoptan disposiciones complementarias para los medicamentos
homeopáticos(13), han sido modificadas en diversas ocasiones y de forma
sustancial. Conviene, en aras de una mayor racionalidad y claridad, proceder
a la codificación de dichas Directivas reagrupándolas en un texto
único.
(2) Toda regulación en materia de producción, distribución
o utilización de los medicamentos debe tener por objetivo esencial la
salvaguardia de la salud pública.
(3) No obstante, los medios que se utilicen para la consecución de este
objetivo no deben obstaculizar el desarrollo de la industria farmacéutica
ni los intercambios de medicamentos en el seno de la Comunidad.
(4) Las disparidades que presentan determinadas disposiciones nacionales y,
en particular, las disposiciones referentes a los medicamentos, con exclusión
de las sustancias o combinaciones de sustancias que sean alimentos, alimentos
destinados a los animales o productos de higiene, obstaculizan los intercambios
de medicamentos en el seno de la Comunidad y por esta razón, repercuten
directamente en el funcionamiento del mercado interior.
(5) Es importante, en consecuencia, suprimir estos obstáculos, y para
alcanzar este objetivo es necesario proceder a una aproximación de las
disposiciones correspondientes.
(6) Con objeto de reducir las disparidades que subsisten es importante, por
una parte, determinar las reglas relativas al control de los medicamentos y
por otra, precisar las funciones que incumben a las autoridades competentes
de los Estados miembros, con miras a asegurar la observancia de las disposiciones
legales.
(7) Los conceptos de nocividad y de efecto terapéutico sólo pueden
entenderse en su relación recíproca y su significación
relativa está en función del desarrollo de la ciencia y del uso
al que se destine el medicamento. Los documentos e informes que se adjunten
a la solicitud de autorización de comercialización deben demostrar
que el beneficio vinculado a la eficacia supera a los riesgos potenciales.
(8) El establecimiento de normas y protocolos para la realización de
pruebas con los medicamentos constituye un medio eficaz para el control de las
mismas y, por consiguiente, para la protección de la salud pública,
y facilita la circulación de los medicamentos en la medida en que fijen
normas comunes para la realización de la formación de expedientes
y la instrucción de solicitudes.
(9) La experiencia demuestra que conviene concretar aún más los
casos en que, con vistas a la autorización de un medicamento esencialmente
similar a un medicamento autorizado, sea innecesario facilitar los resultados
de las pruebas toxicológicas, farmacológicas, o clínicas,
sin dejar de velar por que no se desfavorezca a las empresas innovadoras.
(10) No obstante, a la repetición sin imperiosa necesidad de las pruebas
en personas o animales se oponen consideraciones de orden público.
(11) La adopción de idénticas normas y protocolos por los Estados
miembros permitirá a las autoridades competentes pronunciarse sobre la
base de pruebas uniformes y en función de criterios comunes, contribuyendo
consecuentemente a evitar posibles divergencias en la apreciación.
(12) Con la excepción de los medicamentos sujetos al procedimiento centralizado
de autorización de la Comunidad, previsto en el Reglamento (CEE) n°
2309/93 del Consejo, de 22 de julio de 1993, por el que se establecen procedimientos
comunitarios para la autorización y supervisión de medicamentos
de uso humano y veterinario y por el que se crea la Agencia Europea para la
Evaluación de Medicamentos(14), las autorizaciones de comercialización
otorgadas por la autoridad competente de un Estado miembro han de ser aceptadas,
por las autoridades competentes de los demás Estados miembros, a no ser
que existan motivos graves para suponer que la autorización del medicamento
puede presentar un riesgo para la salud pública. En caso de discrepancias
entre Estados miembros acerca de la calidad, seguridad o eficacia de un medicamento,
debe llevarse a cabo a nivel comunitario una evaluación científica
de la cuestión, que conduzca a una decisión única sobre
los puntos litigiosos, vinculante para los Estados miembros interesados. Tal
decisión debe adoptarse siguiendo un procedimiento rápido que
garantice una estrecha colaboración entre la Comisión y los Estados
miembros.
(13) Con este fin conviene crear un Comité de especialidades farmacéuticas
dependiente de la Agencia Europea para la Evaluación de Medicamentos
establecida por el Reglamento (CEE) n° 2309/93 anteriormente citado.
(14) La presente Directiva constituye una etapa importante en la realización
del objetivo de la libre circulación de los medicamentos. Aún
así pueden resultar necesarias nuevas medidas con el objeto de lograr
este fin, teniendo en cuenta la experiencia adquirida, particularmente en el
seno de dicho Comité de especialidades farmacéuticas, con objeto
de eliminar los obstáculos a la libre circulación que subsisten
todavía.
(15) Para proteger mejor la salud pública y evitar la duplicidad de
esfuerzos mientras se estudien las solicitudes de autorización de comercialización
de los medicamentos, los Estados miembros deben preparar de forma sistemática
informes de evaluación respecto de todos los medicamentos que hayan autorizado,
e intercambiar dichos informes previa solicitud. Además, cualquier Estado
miembro debe poder suspender el estudio de una solicitud de autorización
de comercialización de un medicamento que esté tramitándose
en otro Estado miembro con vistas al reconocimiento de la decisión que
adopte este último Estado miembro.
(16) Tras el establecimiento del mercado interior, sólo podrá
renunciarse a los controles específicos de la calidad de los medicamentos
importados de terceros países cuando la Comunidad haya celebrado los
acuerdos pertinentes para asegurarse de que en el país exportador se
han llevado a cabo los controles necesarios.
(17) Conviene adoptar disposiciones específicas para los medicamentos
inmunológicos, homeopáticos, radiofarmacéuticos, así
como para los medicamentos derivados de la sangre y del plasma humanos.
(18) Cualquier norma relativa a los medicamentos radiofarmacéuticos
debe tener en cuenta lo dispuesto en la Directiva 84/466/Euratom del Consejo,
de 3 de septiembre de 1984, por la que se establecen las medidas fundamentales
relativas a la protección radiológica de las personas sometidas
a exámenes y tratamientos médicos(15). También debe tenerse
en cuenta la Directiva 80/836/Euratom del Consejo, de 15 de julio de 1980, por
la que se modifican las directivas que establecen las normas básicas
relativas a la protección sanitaria de la población y de los trabajadores
contra los peligros que resultan de las radiaciones ionizantes(16) cuyo objetivo
es evitar la exposición de trabajadores o pacientes a radiaciones ionizantes
de nivel excesivo o innecesariamente elevado y, en particular, la letra c) de
su artículo 5, que exige un régimen de autorización previa
para la adición de sustancias radiactivas en la producción y fabricación
de los medicamentos y para la importación de dichos medicamentos.
(19) La Comunidad apoya plenamente los esfuerzos del Consejo de Europa por
fomentar la donación de sangre y de plasma de forma voluntaria y no retribuida
para ir hacia la autosuficiencia en el abastecimiento de productos derivados
de la sangre en toda la Comunidad y para garantizar el respeto de los principios
éticos en el comercio de sustancias terapéuticas de origen humano.
(20) La normativa elaborada para garantizar la calidad, seguridad y eficacia
de los medicamentos derivados de sangre o plasma humanos debe aplicarse de la
misma forma tanto en los establecimientos públicos como en los privados,
así como a la sangre importada de países terceros.
(21) Dadas las especiales características de los medicamentos homeopáticos,
como son la débil concentración de principios activos y las dificultades
para aplicarles la metodología estadística convencional sobre
ensayos clínicos, parece conveniente establecer un procedimiento de registro
simplificado especial para los medicamentos homeopáticos que se comercialicen
sin una indicación terapéutica y en una forma farmacéutica
y dosificación que no presenten riesgo alguno para el paciente.
(22) Los medicamentos antroposóficos descritos en una farmacopea oficial
y preparados según un método homeopático pueden asimilarse,
a efectos de registro y de autorización de comercialización, a
los medicamentos homeopáticos.
(23) Es prioritario indicar muy claramente a quienes utilizan medicamentos
homeopáticos el carácter homeopático de los mismos y ofrecerles
garantías suficientes en cuanto a su calidad y su inocuidad.
(24) Las normas referentes a la fabricación, al control y a las inspecciones
de los medicamentos homeopáticos deben ser armonizadas, a fin de permitir
la circulación en toda la Comunidad de medicamentos seguros y de buena
calidad.
(25) En el caso de los medicamentos homeopáticos que se comercialicen
con indicación terapéutica o en una presentación que pudiera
originar riesgos, con respecto al efecto terapéutico esperado, deberán
aplicarse las normas habituales para la autorización de comercialización
de los medicamentos. En particular, a los Estados miembros en los que exista
una tradición homeopática se les debe permitir la aplicación
de normas específicas para evaluar los resultados de los ensayos tendentes
a demostrar la seguridad y la eficacia de dichos medicamentos, siempre que tales
normas sean notificadas a la Comisión.
(26) A fin de facilitar la circulación de los medicamentos y de evitar
que los controles efectuados en un Estado miembro se efectúen de nuevo
en otro Estado miembro, procede determinar las condiciones mínimas de
fabricación y de importación de medicamentos procedentes de terceros
países y la concesión de las autorizaciones relativas a éstas.
(27) Interesa que en los Estados miembros, la vigilancia y el control de la
fabricación de los medicamentos estén garantizados por una persona
que responda a unas condiciones mínimas de capacitación profesional.
(28) El fabricante, antes de que se le pueda conceder autorización para
comercializar un medicamento inmunológico o de un medicamento derivado
de sangre o plasma humanos, debe demostrar su capacidad de garantizar de manera
continua la conformidad de los lotes; por lo que respecta a los medicamentos
derivados de sangre o plasma humanos, debe además demostrar su capacidad
de garantizar, en la medida que el estado de la técnica lo permita, la
ausencia de contaminación viral específica.
(29) Es preciso armonizar las condiciones en las que se dispensan los medicamentos
al público.
(30) A tal efecto, toda persona que se desplace por la Comunidad tiene derecho
a llevar consigo una cantidad razonable de medicamentos obtenidos lícitamente
para su uso personal. También debe ser posible que una persona establecida
en un Estado miembro reciba desde otro Estado miembro una cantidad razonable
de medicamentos destinados a su uso personal.
(31) Por otra parte, en virtud del Reglamento (CEE) n° 2309/93, algunos
medicamentos son objeto de una autorización comunitaria de comercialización.
En estas condiciones, es conveniente establecer el régimen jurídico
del despacho de los medicamentos amparados por una autorización comunitaria
de comercialización. En consecuencia, conviene fijar los criterios en
que se basarán las decisiones comunitarias que se adopten.
(32) Conviene, pues, en una primera fase, armonizar los principios de base
aplicables a la clasificación relativa al despacho de medicamentos en
la Comunidad o en el Estado miembro interesado, inspirándose en los principios
ya establecidos en la materia por el Consejo de Europa y en los trabajos de
armonización realizados en el marco de las Naciones Unidas en relación
con los estupefacientes y los psicotropos.
(33) Las disposiciones referidas a la clasificación en materia de dispensación
de medicamentos no afectan a las disposiciones de los regímenes nacionales
de seguridad social relativas al reembolso o al pago de los medicamentos sujetos
a receta médica.
(34) Muchas operaciones de distribución al por mayor de medicamentos
para uso humano pueden abarcar simultáneamente varios Estados miembros.
(35) Procede controlar el conjunto de la cadena de distribución de medicamentos,
desde su fabricación o su importación en la Comunidad hasta su
despacho al público, de forma que quede garantizado que los medicamentos
se conservan, transportan y manipulan en condiciones adecuadas. Las disposiciones
que conviene adoptar con este objetivo facilitarán considerablemente
la retirada del mercado de productos defectuosos y permitirán luchar
más eficazmente contra las imitaciones fraudulentas.
(36) Cualquier persona que participe en la distribución al por mayor
de medicamentos debe ser titular de una autorización específica.
No obstante, conviene dispensar de dicha autorización a los farmacéuticos
y personas habilitadas a despachar medicamentos al público y que se limiten
a esta actividad. Sin embargo, para controlar el conjunto de la cadena de distribución
de medicamentos, es necesario que los farmacéuticos y personas facultadas
para dispensar medicamentos al público conserven registros en los que
se indiquen los movimientos de entrada.
(37) La autorización debe someterse a ciertos requisitos esenciales,
cuyo cumplimiento debe verificar el Estado miembro interesado. Cada Estado miembro
debe reconocer las autorizaciones concedidas por los demás Estados miembros.
(38) Determinados Estados miembros imponen a los mayoristas que suministran
medicamentos a los farmacéuticos y a las personas autorizadas a dispensar
medicamentos al público determinadas obligaciones de servicio público.
Dichos Estados miembros deben poder seguir imponiendo dichas obligaciones a
los mayoristas establecidos en su territorio. Asimismo, deben poder aplicarlas
a los mayoristas de los demás Estados miembros siempre que no impongan
ninguna obligación más estricta que las que imponen a sus propios
mayoristas y que puedan considerarse justificadas por razones de protección
de la salud pública y proporcionadas al objetivo relativo a dicha protección.
(39) Es conveniente precisar las normas a las que debe ajustarse el etiquetado
y la redacción del prospecto.
(40) Las disposiciones relativas a la información a los pacientes deben
garantizar un nivel elevado de protección de los consumidores, para permitir
el uso correcto de los medicamentos a partir de una información completa
y comprensible.
(41) La comercialización de los medicamentos cuyo etiquetado y cuyo
prospecto estén realizados con arreglo a lo dispuesto en la presente
Directiva no debe prohibirse ni obstaculizarse por motivos relacionados con
el etiquetado o el prospecto.
(42) La presente Directiva debe entenderse sin perjuicio de la aplicación
de las medidas adoptadas en virtud de la Directiva 84/450/CEE del Consejo, de
10 de septiembre de 1984, relativa a la aproximación de las disposiciones
legales, reglamentarias y administrativas de los Estados miembros en materia
de publicidad engañosa(17).
(43) Todos los Estados miembros han adoptado, además, medidas específicas
relativas a la publicidad de los medicamentos. Entre dichas medidas existen
disparidades. Estas disparidades influyen en el funcionamiento del mercado interior,
por el hecho de que la publicidad que se difunde en un Estado miembro puede
producir efectos en los demás Estados miembros.
(44) La Directiva 89/552/CEE del Consejo, de 3 de octubre de 1989, sobre la
coordinación de determinadas disposiciones legales, reglamentarias y
administrativas de los Estados miembros relativas al ejercicio de actividades
de radiodifusión televisiva(18), prohíbe la publicidad televisada
de medicamentos disponibles únicamente por prescripción facultativa
en el Estado miembro del que dependa el organismo de radiodifusión televisiva.
Procede generalizar este principio ampliándolo a otros medios de comunicación.
(45) La publicidad destinada al público de aquellos medicamentos que
pueden ser dispensados sin prescripción médica podría afectar
a la salud pública, si dicha publicidad fuese excesiva e imprudente.
Esta publicidad, cuando esté permitida, debe, por lo tanto, responder
a determinados criterios esenciales que conviene definir.
(46) Por otro lado, debe prohibirse la distribución gratuita de muestras
al público practicada con fines de promoción.
(47) La publicidad de medicamentos destinada a personas facultadas para prescribirlos
o dispensarlos contribuye a la información de dichas personas. No obstante,
conviene establecer para esta información unas condiciones estrictas
y un control efectivo, inspirándose, principalmente, en los trabajos
realizados en el marco del Consejo de Europa.
(48) La publicidad de medicamentos debe estar sometida a un control adecuado
y eficaz. A este respecto, es conveniente inspirarse en los mecanismos de control
establecidos por la Directiva 84/450/CEE.
(49) Los visitadores médicos desempeñan un papel importante en
la promoción de los medicamentos. Por este motivo, conviene imponerles
determinadas obligaciones, y, en concreto, la obligación de entregar
a la persona que visiten un resumen de las características del producto.
(50) Las personas facultadas para prescribir medicamentos deben realizar esta
tarea de modo totalmente objetivo y sin hallarse influidos por incitaciones
económicas, directas o indirectas.
(51) Conviene poder suministrar muestras gratuitas de medicamentos, respetando
determinadas condiciones restrictivas, a las personas facultadas para prescribirlos
o dispensarlos, con el fin de que se familiaricen con los nuevos medicamentos
y adquieran experiencia respecto a su utilización.
(52) Si bien es importante que las personas facultadas para prescribir o dispensar
medicamentos dispongan de fuentes de información neutrales y objetivas
sobre los medicamentos disponibles en el mercado, es a los Estados miembros,
no obstante, a los que incumbe tomar las medidas oportunas a este fin, en función
de su situación particular.
(53) Conviene que cada empresa fabricante o importadora de medicamentos instaure
un mecanismo mediante el cual pueda garantizarse que toda la información
comunicada respecto a un medicamento se ajusta a las condiciones de uso aprobadas.
(54) A fin de garantizar la seguridad de los medicamentos tras su comercialización,
los sistemas de farmacovigilancia en la Comunidad deben adaptarse permanentemente
al progreso científico y técnico.
(55) Es necesario tener en cuenta las modificaciones resultantes de la armonización
internacional de las definiciones, de la terminología y del desarrollo
tecnológico en el ámbito de la farmacovigilancia.
(56) Con el uso creciente de las redes electrónicas para comunicar información
sobre reacciones adversas a los medicamentos comercializados en la Comunidad
se pretende hacer posible que las autoridades competentes compartan la información
al mismo tiempo.
(57) Interesa a la Comunidad velar por la coherencia de los sistemas de farmacovigilancia
de, por una parte, los medicamentos objeto de un proceso autorización
centralizada y, por otra, de los autorizados por otros procedimientos.
(58) Los titulares de autorizaciones de comercialización deben ser responsables,
respecto de los medicamentos que comercialicen, de una farmacovigilancia continua
y centrada en la prevención.
(59) Procede aprobar las medidas necesarias para la ejecución de la
presente Directiva con arreglo a la Decisión 1999/468/CE del Consejo,
de 28 de junio de 1999, por la que se establecen los procedimientos para el
ejercicio de las competencias de ejecución atribuidas a la Comisión(19).
(60) Es necesario atribuir a la Comisión la facultad de adoptar cualquier
cambio necesario en el Anexo I con el fin de adaptarlo al progreso científico
y técnico.
(61) La presente Directiva no debe afectar a las obligaciones de los Estados
miembros relativas a los plazos de transposición de las Directivas indicadas
en la parte B del Anexo II,
HAN ADOPTADO LA PRESENTE DIRECTIVA:
TÍTULO I
DEFINICIONES
Artículo 1
A efectos de la presente Directiva, se entenderá por:
1) Especialidad farmacéutica:
todo medicamento previamente elaborado, comercializado bajo una denominación
especial y un determinado acondicionamiento;
2) Medicamento:
toda sustancia o combinación de sustancias que se presente como poseedora
de propiedades curativas o preventivas con respecto a las enfermedades humanas;
se considerarán asimismo medicamentos todas las sustancias o combinación
de sustancias que puedan administrarse al hombre con el fin de establecer un
diagnóstico médico o de restablecer, corregir o modificar las
funciones fisiológicas del hombre;
3) Sustancia:
cualquier materia, sin distinción de origen, pudiendo ser éste:
- humano, como:
la sangre humana y sus productos derivados;
- animal, como:
los microorganismos, animales enteros, partes de órganos, secreciones
animales, toxinas, sustancias obtenidas por extracción, productos derivados
de la sangre;
- vegetal, como:
los microorganismos, plantas, partes de plantas, secreciones vegetales, sustancias
obtenidas por extracción;
- químico, como:
los elementos, materias químicas naturales y productos químicos
de transformación y de síntesis;
4) Medicamento inmunológico:
todo medicamento consistente en vacunas, toxinas, sueros y alergenos:
a) las vacunas, toxinas o sueros, que comprenden en particular:
i) los agentes utilizados para provocar una inmunidad activa como la vacuna
anticolérica, el BCG, la vacuna antipoliomelítica, la vacuna antivariólica;
ii) los agentes utilizados para diagnosticar el estado de inmunidad, en particular
la tuberculina y la tuberculina PPD, las toxinas utilizadas en los tests de
Schick y de Dick, la brucelina;
iii) los agentes utilizados para provocar una inmunidad pasiva, como la antitoxina
diftérica, la globulina antivariólica, la globulina antilinfocítica;
b) los productos alergénicos comprendiendo cualquier medicamento destinado
a detectar o provocar una alteración adquirida y específica en
la respuesta inmunológica a un agente alergizante;
5) Medicamento homeopático:
todo medicamento obtenido a partir de productos, sustancias o compuestos denominados
cepas homeopáticas, con arreglo a un procedimiento de fabricación
homeopático descrito en la Farmacopea europea o, en su defecto, en las
farmacopeas utilizadas en la actualidad de forma oficial en los Estados miembros;
un medicamento homeopático podrá igualmente contener varios principios;
6) Radiofármaco:
cualquier medicamento que, cuando esté preparado para su uso, contenga
uno o más radionucleidos (isótopos radioactivos), incorporados
con algún objetivo médico;
7) Generador de radionucleidos:
cualquier sistema que incorpore un radionucleido padre fijo a partir del cual
se produzca un radionucleido hijo obtenido por elución o por cualquier
otro método y utilizado en un radiofármaco;
8) Equipo reactivo de radionucleidos:
cualquier preparado que deba reconstituirse o combinarse con radionucleidos
en radiofármaco final, generalmente antes de su administración;
9) Precursor de radionucleidos:
cualquier otro radionucleido producido para el marcado radiactivo de otras sustancias
antes de su administración;
10) Medicamentos derivados de sangre o plasma humanos:
medicamentos a base de constituyentes sanguíneos preparados industrialmente
por establecimientos públicos o privados; dichos medicamentos comprenden,
en particular, albúmina, factores de coagulación e inmunoglobulinas
de origen humano;
11) Reacción adversa:
cualquier respuesta nociva e involuntaria a un medicamento, producida a dosis
aplicadas normalmente en el hombre para la profilaxis, el diagnóstico
o el tratamiento de enfermedades, o para el restablecimiento, la corrección
o la modificación de funciones fisiológicas;
12) Reacción adversa grave:
cualquier reacción adversa que ocasione la muerte, ponga en peligro la
vida del paciente, exija una hospitalización o la prolongación
de la misma, ocasione una invalidez o una incapacidad significativa o persistente
o produzca una anomalía o malformación congénita;
13) Reacción adversa inesperada:
cualquier reacción adversa cuya naturaleza, gravedad o consecuencias
no concuerden con el resumen de las características del producto;
14) Informes periódicos de actualizados en materia de seguridad:
los informes periódicos que contengan las informaciones registradas con
arreglo al artículo 104;
15) Estudios de seguridad posteriores a la autorización:
un estudio farmacoepidemiológico o un ensayo clínico efectuados
de conformidad con los términos de la autorización de comercialización
y realizado con el propósito de identificar o cuantificar un riesgo para
la seguridad relativo a un medicamento autorizado;
16) Abuso de medicamentos:
el uso excesivo intencionado, persistente o esporádico, de medicamentos,
acompañado de reacciones nocivas físicas o psicológicas;
17) Distribución al por mayor de medicamentos:
toda actividad que consista en obtener, conservar, suministrar o exportar medicamentos,
excluido el despacho de medicamentos al público; estas actividades serán
realizadas con fabricantes o sus depositarios, importadores, otros mayoristas
o con los farmacéuticos y personas autorizadas o facultadas, en el Estado
miembro de que se trate, para dispensar medicamentos al público;
18) Obligación de servicio público:
la obligación de los mayoristas de garantizar permanentemente una provisión
de medicamentos suficiente para responder a las necesidades de un territorio
geográficamente determinado, así como la entrega del suministro
solicitado, en cualquier punto de ese territorio, en plazos muy breves;
19) Receta médica:
cualquier receta médica extendida por un profesional de la salud facultado
a estos efectos;
20) Denominación del medicamento:
la denominación, que podrá ser un nombre arbitrario o una denominación
común o científica acompañada de una marca o del nombre
del fabricante; cuando sea un nombre arbitrario, no deberá prestarse
a confusión con la denominación común;
21) Denominación común:
la denominación común internacional recomendada por la Organización
Mundial de la Salud o, en su defecto, la denominación común usual;
22) Dosis del medicamento:
el contenido de substancia activa, expresado en cantidad por unidad de toma,
por unidad de volumen o de peso en función de la presentación;
23) Acondicionamiento primario:
el envase o cualquier otra forma de acondicionamiento que se encuentre en contacto
directo con el medicamento;
24) Embalaje exterior:
el embalaje en que se encuentre el acondicionamiento primario;
25) Etiquetado:
las menciones que consten en el embalaje exterior o en el acondicionamiento
primario;
26) Prospecto:
la nota informativa para el usuario, que acompañe al medicamento;
27) Agencia:
la Agencia Europea para la Evaluación de Medicamentos establecida por
el Reglamento (CEE) n° 2309/93.
28) Riesgo para la salud pública:
todo riesgo referido a la calidad, la seguridad y la eficacia del medicamento.
TÍTULO II
ÁMBITO DE APLICACIÓN
Artículo 2
Las disposiciones de la presente Directiva se aplicarán a los medicamentos para uso humano producidos industrialmente y destinados a ser comercializados en los Estados miembros.
Artículo 3
La presente Directiva no se aplicará a:
1) los medicamentos preparados en una farmacia de acuerdo con una prescripción
médica destinada a un enfermo determinado (denominada comúnmente
fórmula magistral);
2) los medicamentos preparados en una farmacia de acuerdo con las indicaciones
de una farmacopea, destinados a su dispensación directa a los enfermos
a los que abastece dicha farmacia (denominada comúnmente fórmula
oficinal);
3) los medicamentos destinados a pruebas de investigación y de desarrollo;
4) los productos intermedios destinados a una transformación posterior
realizada por un fabricante autorizado;
5) los radionucleidos en forma de fuentes selladas;
6) la sangre completa, al plasma ni a las células sanguíneas de
origen humano.
Artículo 4
1. Ninguna disposición de la presente Directiva dispensará del
cumplimiento de la normativa comunitaria sobre protección contra la radiación
de personas sometidas a exámenes o tratamientos médicos ni de
la normativa comunitaria que establece las normas básicas de seguridad
para la protección de la salud pública y de los trabajadores contra
los peligros de la radiación ionizante.
2. La presente Directiva no afectará a la Decisión 86/346/CEE
del Consejo, de 25 de junio de 1986, por la que se acepta, en nombre de la Comunidad,
el Acuerdo europeo relativo al intercambio de sustancias terapéuticas
de origen humano(20).
3. Las disposiciones de la presente Directiva no afectarán a las competencias
de los Estados miembros, ni en lo que respecta a la fijación de los precios
de los medicamentos ni a su inclusión en el ámbito de aplicación
de los sistemas nacionales de seguridad social, por motivos sanitarios, económicos
y sociales.
4. La presente Directiva no afectará a la aplicación de las legislaciones
nacionales que prohíban o restrinjan la venta, el suministro o el uso
de medicamentos con fines contraceptivos o abortivos. Los Estados miembros comunicarán
a la Comisión las disposiciones nacionales correspondientes.
Artículo 5
Los Estados miembros podrán, de acuerdo con la legislación vigente y con miras a atender necesidades especiales, excluir de las disposiciones de la presente Directiva a los medicamentos que se suministren atendiendo a un encargo leal y no solicitado, elaborados de acuerdo con las especificaciones de un facultativo reconocido y que los destine a sus pacientes particulares bajo su responsabilidad personal directa.
TÍTULO III
COMERCIALIZACIÓN
CAPÍTULO 1
Autorización de comercialización
Artículo 6
1. No podrá comercializarse ningún medicamento en un Estado miembro
sin que la autoridad competente de dicho Estado miembro haya concedido una autorización
de comercialización, de conformidad con la presente Directiva, o sin
que se haya concedido una autorización de conformidad con lo dispuesto
en el Reglamento (CEE) n° 2309/93.
2. La autorización mencionada en el apartado 1 será asimismo exigida
para los generadores de radionucleidos, equipos reactivos de radionucleidos
y los radiofármacos precursores de radionucleidos, así como para
los radiofármacos preparados industrialmente.
Artículo 7
Sin embargo no se exigirá autorización para un radiofármaco preparado en el momento de su uso por una persona o una institución que según la legislación nacional estén autorizadas a utilizar estos medicamentos, en un centro asistencial autorizado, exclusivamente a partir de generadores de radionucleidos, equipos reactivos de radionucleidos o precursores de radionucleidos autorizados, con arreglo a las instrucciones del fabricante.
Artículo 8
1. Con objeto de lograr la autorización de comercialización de
un medicamento no prevista en el procedimiento establecido por el Reglamento
(CEE) n° 2309/93, se presentará una solicitud ante la autoridad competente
del Estado miembro en cuestión.
2. La autorización de comercialización sólo podrá
concederse a aquellos solicitantes que se encuentren establecidos en la Comunidad.
3. La solicitud irá acompañada de los siguientes datos y documentos
presentados, con arreglo al Anexo I:
a) nombre o razón social y domicilio o sede social del solicitante y,
en su caso, del fabricante;
b) denominación del medicamento;
c) composición cualitativa y cuantitativa de todos los componentes del
medicamento, de acuerdo con la terminología ordinaria, excluyendo las
fórmulas químicas empíricas, y con la denominación
común internacional recomendada por la Organización Mundial de
la Salud, cuando exista tal denominación;
d) descripción del modo de fabricación;
e) indicaciones terapéuticas, contraindicaciones y reacciones adversas;
f) posología, forma farmacéutica, modo y vía de administración
y período o plazo de validez previsto;
g) si ha lugar, las razones por las cuales han de tomarse medidas de precaución
o de seguridad al almacenar el medicamento, al administrarlo a los pacientes
y al eliminar los productos residuales, junto con la indicación de cualquier
riesgo potencial que presente el medicamento para el medio ambiente;
h) descripción de los métodos de control utilizados por el fabricante
(análisis cualitativo y cuantitativo de los componentes y del producto
acabado, pruebas especiales, por ejemplo, pruebas de esterilidad, pruebas para
la búsqueda de sustancias pirógenas, búsqueda de metales
pesados, pruebas de estabilidad, pruebas biológicas y de toxicidad, controles
de los productos intermedios de la fabricación);
i) resultado de las pruebas:
- fisioquímicas, biológicas o microbiológicas;
- toxicológicas y farmacológicas;
- clínicas;
j) un resumen de las características del producto, con arreglo al artículo
11, una o varias muestras o maquetas del embalaje exterior y del acondicionamiento
primario del medicamento así como el prospecto;
k) un documento del que se desprenda que el fabricante está autorizado
en su país para fabricar medicamentos;
l) una copia de la autorización de comercialización obtenida para
el medicamento en otro Estado miembro o en un tercer país, junto con
la lista de los Estados miembros en los que se esté estudiando una solicitud
de autorización presentada de conformidad con la presente Directiva,
una copia del resumen de las características del producto propuesto por
el solicitante en virtud del artículo 11 o aprobado por las autoridades
competentes del Estado miembro en virtud del artículo 21, y una copia
del prospecto propuesto con arreglo al artículo 59 o aprobado por la
autoridad competente del Estado miembro de conformidad con el artículo
61; los detalles de cualquier decisión de denegación de autorización,
tanto en la Comunidad como en un país tercero, y los motivos de tal decisión.
Esta información deberá actualizarse periódicamente.
Artículo 9
Además de los requisitos establecidos en el artículo 8 y en el
apartado 1 del artículo 10, las solicitudes de autorización de
comercialización de un generador de radionucleidos incluirán también
la información y las características siguientes:
- una descripción general del sistema junto con una descripción
detallada de los componentes del mismo que puedan afectar a la composición
o calidad del preparado nucleido hijo;
- características cualitativas y cuantitativas del eluido o sublimado.
Artículo 10
1. No obstante lo dispuesto en la letra i) del apartado 3 del artículo
8, y sin perjuicio del derecho relativo a la protección de la propiedad
industrial y comercial:
a) el solicitante no tendrá obligación de facilitar los resultados
de las pruebas toxicológicas y farmacológicas o los de las pruebas
clínicas, si puede demostrar:
i) bien que el medicamento es esencialmente similar a un medicamento autorizado
en el Estado miembro para el que se curse la solicitud y que el titular de la
autorización de comercialización del medicamento original consiente
en que, para el estudio de la solicitud de que se trate, se haga uso de la documentación
toxicológica, farmacológica y/o clínica que obra en el
expediente del medicamento original;
ii) bien, que el o los componentes del medicamento tienen un uso médico
claramente establecido y presentan una eficacia reconocida así como un
nivel aceptable de seguridad, mediante una bibliografía científica
detallada;
iii) bien que el medicamento es esencialmente similar a algún otro medicamento
autorizado en la Comunidad, según las disposiciones comunitarias vigentes,
desde hace seis años como mínimo y comercializado en el Estado
miembro para el que se curse la solicitud; el citado período se elevará
a diez años cuando se trate de medicamentos de alta tecnología
autorizados de acuerdo con el procedimiento establecido por el apartado 5 del
artículo 2 de la Directiva 87/22/CEE del Consejo(21); además,
los Estados miembros podrán igualmente ampliar el citado período
a diez años, mediante una decisión única que cubra todos
los medicamentos comercializados en sus respectivos territorios, cuando estimen
que así lo exigen las necesidades de la salud pública. Los Estados
miembros podrán suspender la aplicación del período de
seis años una vez pasada la fecha del agotamiento de la patente que ampare
el medicamento original.
Sin embargo, en los casos en que el medicamento esté destinado a un uso
terapéutico diferente o deba administrarse por vías diferentes
o con dosificación diferente con respecto a los otros medicamentos comercializados,
deberán suministrarse los resultados de las pruebas toxicológicas,
farmacológicas y/o clínicas apropiadas.
b) por lo que se refiere a los nuevos medicamentos que contengan componentes
conocidos, que no hayan sido combinados todavía con fines terapéuticos,
deberán facilitarse los resultados de las pruebas toxicológicas,
farmacológicas y clínicas relativas a la combinación, sin
necesidad de facilitar la documentación relativa a cada componente individual.
2. El Anexo I se aplicará por analogía, cuando se presente una
bibliografía científica detallada en virtud del inciso ii) de
la letra a) del apartado 1.
Artículo 11
El resumen de las características del producto recogerá las informaciones
siguientes:
1) denominación del medicamento;
2) composición cualitativa y cuantitativa en sustancias activas y en
componentes del excipiente cuyo conocimiento sea necesario para una buena administración
del medicamento; se emplearán las denominaciones comunes o las denominaciones
químicas;
3) forma farmacéutica;
4) propiedades farmacológicas y, en la medida en que dichas informaciones
sean útiles para la utilización terapéutica, elementos
de farmacocinética;
5) informaciones clínicas:
5.1. informaciones terapéuticas,
5.2. contraindicaciones,
5.3. reacciones adversas (frecuencia y gravedad),
5.4. precauciones particulares de empleo, y para los medicamentos inmunológicos,
deberán tomarse precauciones especiales por parte de las personas que
manipulan el medicamento inmunológico y lo administran a los pacientes,
y, en su caso, el paciente deberá tomar precauciones,
5.5. utilización en caso de embarazo y lactancia,
5.6. interacciones con otros medicamentos y otras formas de interacción,
5.7. posología y modo de administración para adultos y, en la
medida en que ello sea necesario, para niños,
5.8. sobredosis (síntomas, socorros de urgencia, antídotos),
5.9. advertencias especiales,
5.10. efectos sobre la capacidad de conducir y de usar máquinas;
6) informaciones farmacéuticas:
6.1. incompatibilidades mayores,
6.2. duración, si fuera necesario tras la reconstitución del medicamento
o cuando el acondicionamiento primario sea abierto por primera vez,
6.3. precauciones particulares de conservación,
6.4. naturaleza y contenido del acondicionamiento primario,
6.5. precauciones especiales para eliminar medicamentos no utilizados o en su
caso, residuos derivados de dichos medicamentos;
7) nombre o razón social y domicilio o sede social del titular de la
autorización de comercialización;
8) para los radiofármacos, explicación detallada completa de la
dosimetría interna de la radiación;
9) para los radiofármacos, instrucciones detalladas suplementarias para
la preparación extemporánea y el control de calidad de esta preparación
y, en su caso, tiempo máximo de almacenamiento durante el cual, cualquier
preparado intermedio, como un eluido, o el radiofármaco listo para su
empleo cumplan las especificaciones previstas.
Artículo 12
1. Los Estados miembros adoptarán todas las disposiciones adecuadas
para que los documentos e informes enumerados en las letras h) e i) del apartado
3 del artículo 8 y en el inciso ii) de la letra a) del apartado 1 del
artículo 10 sean establecidos por expertos que reúnan las cualificaciones
técnicas o profesionales necesarias, antes de ser presentados a las autoridades
competentes. Estos documentos e informes serán firmados por dichos expertos.
2. Según sus cualificaciones, la función de tales expertos será:
a) efectuar los trabajos relacionados con su disciplina (análisis, farmacología
y ciencias experimentales análogas, clínica) y describir objetivamente
los resultados obtenidos (cuantitativos y cualitativos);
b) describir las comprobaciones que hayan realizado con arreglo al Anexo I,
así como el de dictaminar especialmente:
- en el caso del analista, si el medicamento guarda conformidad con la composición
declarada, proporcionando para ello todas las justificaciones pertinentes en
relación con los métodos de control que utilizará el fabricante,
- en el caso del farmacólogo o en el del especialista de análoga
competencia experimental, cuál es la toxicidad del medicamento y cuáles
son sus propiedades farmacológicas comprobadas,
- en el caso del clínico, si ha podido encontrar en las personas tratadas
con el medicamento los efectos correspondientes a los informes aportados por
el solicitante en aplicación de los artículos 8 y 10 y, si el
producto medicamento se tolera bien, qué posología aconseja y
cuáles son las eventuales contraindicaciones y reacciones adversas;
c) justificar el recurso eventual a la bibliografía científica
detallada mencionada en el inciso ii) de la letra a) del apartado 1 del artículo
10, en las condiciones previstas en el Anexo I.
3. Los informes detallados de los expertos formarán parte del expediente
que el solicitante presente a las autoridades competentes.
CAPÍTULO 2
Disposiciones particulares aplicables a los medicamentos homeopáticos
Artículo 13
1. Los Estados miembros velarán para que los medicamentos homeopáticos
que se fabriquen y comercialicen en la Comunidad se registren o autoricen con
arreglo a lo dispuesto en los artículos 14, 15 y 16, excepto cuando dichos
medicamentos estén cubiertos por un registro o por una autorización
acordado(a) de conformidad con la legislación nacional hasta el 31 de
diciembre de 1993 (e independientemente de la extensión de dicho registro
o de dicha autorización después de esa fecha). Cada Estado miembro
tendrá debidamente en cuenta los registros o las autorizaciones ya expedidos
por otro Estado miembro.
2. Un Estado miembro podrá abstenerse de establecer un procedimiento
de registro simplificado especial de los medicamentos homeopáticos a
que se refiere el artículo 14. El Estado miembro informará de
ello a la Comisión. Dicho Estado miembro deberá permitir, en tal
caso, la utilización en su territorio de los medicamentos registrados
por otros Estados miembros con arreglo a los artículos 14 y 15.
Artículo 14
1. Sólo podrán acogerse a un procedimiento de registro simplificado
especial los medicamentos homeopáticos que cumplan todas las condiciones
que a continuación se exponen:
- vía de administración oral o externa;
- ausencia de indicación terapéutica particular en la etiqueta
o en cualquier información relativa al medicamento;
- grado de dilución que garantice la inocuidad del medicamento; en particular,
el preparado no deberá contener más de una parte por 10000 de
tintura madre ni más de la centésima parte de la dosis más
baja que eventualmente se emplee en medicina alopática de aquellas sustancias
activas cuya presencia en un medicamento alopático implique la obligatoriedad
de presentar receta médica.
Los Estados miembros llevarán a cabo la clasificación relativa
a la dispensación del medicamento en el momento del registro.
2. Los criterios y las normas de procedimiento previstos en el apartado 4 del
artículo 4, en el apartado 1 del artículo 17 y en los artículos
22 a 26, 112, 116 y 125 serán aplicables por analogía al procedimiento
de registro simplificado especial de los medicamentos homeopáticos, con
excepción de la prueba del efecto terapéutico.
3. La prueba del efecto terapéutico no será necesaria para los
medicamentos homeopáticos registrados con arreglo al apartado 1 del presente
artículo, o, en su caso, admitidos según el apartado 2 del artículo
13.
Artículo 15
La solicitud de registro simplificada especial podrá abarcar toda una
serie de medicamentos obtenidos a partir de la(s) misma(s) cepa(s) homeopática(s).
A dicha solicitud se adjuntarán los documentos siguientes, a fin de demostrar,
principalmente, la calidad farmacéutica y la homogeneidad de los lotes
de fabricación de dichos medicamentos:
- denominación científica u otra denominación que figure
en una farmacopea, de la(s) cepa(s) homeopática(s), con mención
de las distintas vías de administración, formas farmacéuticas
y grados de dilución que vayan a registrarse;
- informe en el que se describa la obtención y el control de la cepa
o cepas y se justifique su carácter homeopático, basándose
en una bibliografía apropiada;
- informe sobre la fabricación y el control de cada una de las formas
farmacéuticas, acompañado de una descripción de los métodos
de dilución y de dinamización;
- autorización para fabricar los medicamentos en cuestión;
- copia de los registros o autorizaciones eventualmente obtenidos para esos
mismos medicamentos en otros Estados miembros;
- una o varias muestras o maquetas del embalaje exterior y del acondicionamiento
primario de los medicamentos que vayan a registrarse;
- información sobre la estabilidad del medicamento.
Artículo 16
1. Los medicamentos homeopáticos distintos de los contemplados en el
apartado 1 del artículo 14 serán autorizados y etiquetados con
arreglo a los artículos 8, 10 y 11.
2. Los Estados miembros podrán introducir o mantener en sus respectivos
territorios normas específicas para las pruebas toxicológicas,
farmacológicas y clínicas de los medicamentos homeopáticos
distintos de los contemplados en el apartado 1 del artículo 14, con arreglo
a los principios y particularidades de la medicina homeopática de cada
Estado miembro.
En ese caso, el Estado miembro de que se trate notificará a la Comisión
las normas específicas vigentes.
3. Las disposiciones del título IX serán aplicables además
a los medicamentos homeopáticos, con excepción de los contemplados
en el apartado 1 del artículo 14.
CAPÍTULO 3
Procedimiento relativo a la autorización de comercialización
Artículo 17
1. Los Estados miembros adoptarán todas las medidas adecuadas para garantizar
que el procedimiento para conceder una autorización de comercialización
de un medicamento se haya efectuado dentro de los doscientos diez días
siguientes a la presentación de una solicitud válida.
2. Si un Estado miembro tuviere conocimiento de que en otro Estado miembro se
está estudiando de forma efectiva una solicitud de autorización
de un medicamento podrá dejar en suspenso el estudio detallado de la
solicitud a la espera del informe de evaluación preparado por el otro
Estado miembro de conformidad con el apartado 4 del artículo 21.
El Estado miembro interesado informará al otro Estado miembro y al solicitante
acerca de su decisión de dejar en suspenso el examen minucioso de dicha
solicitud. Tan pronto como haya terminado el examen de la solicitud y haya tomado
una decisión, el otro Estado miembro enviará una copia de su informe
de evaluación al Estado miembro interesado.
Artículo 18
Si se informare a un Estado miembro, de conformidad con lo dispuesto en la
letra l) del apartado 3 del artículo 8, de que otro Estado miembro ha
autorizado un medicamento para el que se ha solicitado una autorización
en el Estado miembro interesado, dicho Estado miembro solicitará sin
demora a las autoridades del Estado miembro que hubiere concedido la autorización
que le envíen el informe de evaluación a que se refiere el apartado
4 del artículo 21.
Dentro de los 90 días siguientes a la recepción del informe de
evaluación, el Estado miembro interesado deberá reconocer la decisión
del primer Estado miembro y el resumen de las características del medicamento
que éste hubiera aprobado o, si considera que existen motivos para suponer
que la autorización del medicamento de que se trate puede constituir
un riesgo para la salud pública, aplicar los procedimientos contemplados
en los artículos 29 a 34.
Artículo 19
Para instruir la solicitud presentada en virtud del artículo 8 y del
apartado 1 del artículo 10, la autoridad competente de los Estados miembros:
1) deberá verificar la conformidad del expediente presentado con el artículo
8 y el apartado 1 del artículo 10 y examinará si reúne
las condiciones de concesión de la autorización de comercialización;
2) podrá someter el medicamento, sus materias primas y de ser necesario,
sus productos intermedios u otros componentes, al control de un laboratorio
estatal o de un laboratorio designado a este efecto y se asegura de que son
satisfactorios los métodos de control utilizados por el fabricante y
descritos en la documentación de la solicitud, con arreglo a la letra
h) del apartado 3 del artículo 8;
3) podrá, en su caso, exigir del solicitante que complete el expediente
en lo referente a los elementos mencionados en el apartado 3 del artículo
8 y en el apartado 1 del artículo 10. Cuando la autoridad competente
haga uso de esta facultad, los plazos previstos en el artículo 17 quedarán
suspendidos hasta que se proporcionen los datos complementarios requeridos.
Asimismo, dichos plazos quedarán suspendidos durante el tiempo concedido,
en su caso, al solicitante para que se explique oralmente o por escrito.
Artículo 20
Los Estados miembros adoptarán todas las disposiciones adecuadas a fin
de que:
a) las autoridades competentes verifiquen que los fabricantes e importadores
de medicamentos procedentes de terceros países estén capacitados
para llevar a cabo la fabricación en observancia de las indicaciones
facilitadas para la aplicación de la letra d) del apartado 3 del artículo
8 y/o efectuar los controles según los métodos descritos en el
expediente con arreglo a la letra h) del apartado 3 del artículo 8;
b) las autoridades competentes puedan autorizar a los fabricantes e importadores
de medicamentos procedentes de terceros países, en casos excepcionales
y justificados, a que determinadas fases de la fabricación y/o determinados
controles previstos en la letra a), se efectúen por terceros; en este
caso, las verificaciones de las autoridades competentes se efectuarán
igualmente en el establecimiento elegido.
Artículo 21
1. Cuando se conceda una autorización de comercialización, la
autoridad competente del Estado miembro interesado comunicará al titular
de la comercialización del medicamento que aprueba el resumen de las
características del producto.
2. La autoridad competente adoptará todas las medidas necesarias para
que la información que figure en el resumen concuerde con la aprobada
en el momento de la concesión de la autorización o posteriormente.
3. La autoridad competente enviará a la Agencia una copia de la autorización,
junto con el resumen de las características del producto.
4. La autoridad competente elaborará un informe de evaluación
y comentarios sobre el expediente, por lo que se refiere a los resultados de
los ensayos analíticos, farmacotoxicológicos y clínicos
del medicamento de que se trate. El informe de evaluación se actualizará
cuando se disponga de nuevos datos que sean importantes para la evaluación
de la calidad, la seguridad y la eficacia del medicamento.
Artículo 22
En circunstancias excepcionales y previa consulta al solicitante, podrá
concederse una autorización supeditada a determinadas obligaciones específicas
en relación con:
- la realización de estudios complementarios tras la concesión
de la autorización,
- la comunicación de reacciones adversas al medicamento.
Estas decisiones excepcionales sólo podrán tomarse por razones
objetivas y verificables y deberán basarse en uno de los motivos mencionados
en el punto 6 de la Parte II del Anexo I.
Artículo 23
Tras la concesión de una autorización, el titular de la autorización
de la comercialización deberá tener en cuenta, por lo que respecta
a los métodos de fabricación y de control previstos en las letras
d) y h) del apartado 3 del artículo 8, los avances científicos
y técnicos e introducir las modificaciones necesarias para que el medicamento
sea fabricado y controlado según métodos científicos generalmente
aceptados.
Dichas modificaciones deberán ser sometidas a la aprobación de
la autoridad competente del Estado miembro interesado.
Artículo 24
La autorización será válida durante cinco años y podrá renovarse por períodos de cinco años, previa solicitud del titular presentada al menos tres meses antes de la fecha de expiración y previo examen por la autoridad competente de un informe que incluya en particular el estado de la farmacovigilancia y las demás informaciones pertinentes para la vigilancia del medicamento.
Artículo 25
La autorización no afectará a la responsabilidad civil o penal del fabricante y, en su caso, del titular.
Artículo 26
Se denegará la autorización cuando de la comprobación
de los datos y documentos enumerados en el artículo 8 y en el apartado
1 del artículo 10, se desprenda que:
a) el medicamento es nocivo en sus condiciones normales de empleo, o
b) que el medicamento no tiene efecto terapéutico alguno o éste
no ha sido suficientemente justificado por el solicitante, o
c) el medicamento no tiene la composición cualitativa y cuantitativa
declarada.
Se denegará asimismo la autorización cuando la documentación
y los datos que se hubieran presentado como fundamento de la solicitud no se
ajusten a lo dispuesto en el artículo 8 y en el apartado 1 del artículo
10.
CAPÍTULO 4
Reconocimiento mutuo de autorizaciones
Artículo 27
1. Para facilitar la adopción, por parte de los Estados miembros, de
decisiones comunes sobre la autorización de medicamentos, basándose
en criterios científicos de calidad, seguridad y eficacia, y conseguir
así la libre circulación de medicamentos en la Comunidad, se crea
un Comité de especialidades farmacéuticas, denominado en lo sucesivo
"el Comité". El Comité estará adscrito a la Agencia.
2. Además de las otras funciones que le confiera el Derecho comunitario,
el Comité tendrá por misión examinar los asuntos relativos
a la concesión, modificación, suspensión o retirada de
la autorización de comercialización que se hayan presentado de
acuerdo con lo dispuesto en la presente Directiva.
3. El Comité establecerá su propio reglamento interno.
Artículo 28
1. Antes de presentar una solicitud de reconocimiento de una autorización
de comercialización, el titular de la autorización informará
al Estado miembro que haya concedido la autorización en la que se base
la solicitud (en lo sucesivo Estado miembro de referencia), de que se presentará
una solicitud con arreglo a la presente Directiva, y le notificará todas
las adiciones efectuadas en el expediente inicial; dicho Estado miembro podrá
pedir al solicitante toda la información y todos los documentos necesarios
para poder comprobar si los expedientes presentados son idénticos.
Además, el titular de la autorización solicitará al Estado
miembro de referencia que elabore un informe de evaluación sobre el medicamento
en cuestión o, si fuere necesario, que actualice el informe de evaluación,
si éste existe. Dicho Estado miembro elaborará el informe de evaluación
o lo actualizará en un plazo de 90 días a partir de la recepción
de la solicitud.
En el momento en que se presente la solicitud de conformidad con el apartado
2, el Estado miembro de referencia transmitirá el informe de evaluación
al Estado miembro o los Estados miembros afectados por la solicitud.
2. Para que se pueda aceptar en uno o varios Estados miembros una autorización
de comercialización expedida por un Estado miembro de conformidad con
lo dispuesto en el presente Capítulo, el titular de dicha autorización
presentará una solicitud ante las autoridades competentes del Estado
miembro o Estados miembros interesados acompañando la información
y datos a que se refieren el artículo 8, el apartado 1 del artículo
10 y el artículo 11. El titular deberá acreditar que el expediente
es idéntico al aceptado por el Estado miembro de referencia que le concedió
la autorización, o señalar las adiciones o modificaciones que
pueda contener. En ese caso, deberá certificar que el resumen de las
características del producto que él mismo propone, de conformidad
con el artículo 11, es idéntico al aceptado por el Estado miembro
de referencia que le concedió la autorización de conformidad con
el artículo 21. También deberá certificar que todos los
expedientes presentados dentro de este procedimiento son idénticos.
3. El titular de la autorización de comercialización comunicará
a la Agencia la solicitud, le informará de los Estados miembros interesados,
así como de las fechas de presentación de las solicitudes, y le
enviará una copia de la autorización concedida por el Estado miembro
de referencia. Deberá enviar también a la Agencia copias de cualquier
otra autorización de comercialización concedida por los demás
Estados miembros para ese mismo medicamento, y tendrá que indicar si
una solicitud de autorización es ya objeto de examen en un Estado miembro.
4. Salvo en el supuesto excepcional previsto en el apartado 1 del artículo
29, cada Estado miembro aceptará la primera autorización de comercialización
concedida por el Estado miembro de referencia dentro de los 90 días siguientes
a la recepción de la solicitud. Informará de ello al Estado miembro
de referencia que concedió la primera autorización, a los demás
Estados miembros a los que concierna la solicitud, a la Agencia y al titular
de la autorización de comercialización.
Artículo 29
1. Si un Estado miembro considera que existen motivos para pensar que la autorización
de comercialización del medicamento puede presentar un riesgo para la
salud pública, informará de ello sin demora al solicitante, al
Estado miembro de referencia, a los demás Estados miembros implicados
en la solicitud y a la Agencia. El Estado miembro expondrá minuciosamente
sus motivos e indicará las medidas que pueden resultar necesarias para
corregir las insuficiencias de la solicitud.
2. Todos los Estados miembros interesados procurarán llegar a un acuerdo
sobre las medidas que deben adoptarse con respecto a la solicitud. Ofrecerán
al solicitante la posibilidad de exponer su punto de vista oralmente o por escrito.
No obstante, si, en el plazo mencionado en el apartado 4 del artículo
28, los Estados miembros no hubieren llegado a un acuerdo, someterán
sin demora el asunto a la Agencia para que se aplique el procedimiento establecido
en el artículo 32.
3. Dentro del plazo previsto en el apartado 4 del artículo 28, los Estados
miembros interesados proporcionarán al Comité una declaración
pormenorizada de las cuestiones sobre las que no pudieron alcanzar un acuerdo
y los motivos de su desacuerdo. Se facilitará al solicitante una copia
de esta información.
4. En cuanto el solicitante haya sido informado de que se ha recurrido al Comité,
le enviará sin demora una copia de la información y documentos
mencionados en el apartado 2 del artículo 28.
Artículo 30
Cuando un medicamento haya sido objeto de varias solicitudes de comercialización
presentadas de conformidad con el artículo 8, el apartado 1 del artículo
10 y el artículo 11, y los Estados miembros hayan adoptado decisiones
discrepantes en relación con la autorización, o con la suspensión
de ésta o su retirada, cualquier Estado miembro, la Comisión o
el titular de la autorización de comercialización podrán
recurrir al Comité para que se aplique el procedimiento establecido en
el artículo 32.
El Estado miembro interesado, el titular de la autorización de comercialización
o la Comisión deberán identificar con claridad el asunto que se
somete a la consideración del Comité y, si fuere necesario, informarán
de ello al titular.
Los Estados miembros y el titular de la autorización de comercialización
enviarán al Comité toda la información disponible sobre
el asunto.
Artículo 31
En casos específicos en los que estén en juego los intereses
de la Comunidad, los Estados miembros, la Comisión, el solicitante o
el titular de la autorización podrán recurrir al Comité
para que se aplique el procedimiento establecido en el artículo 32 antes
de que se adopte una decisión acerca de una solicitud de autorización
de comercialización, o acerca de la suspensión o revocación
de una autorización, o de cualquier otra modificación necesaria
de los términos en que esté formulada una autorización
de comercialización, especialmente para tener en cuenta la información
recogida de acuerdo con lo dispuesto en el Título IX.
El Estado miembro interesado o la Comisión deberán identificar
con claridad la cuestión que se somete a la consideración del
Comité e informarán de ello al titular de la autorización
de comercialización.
Los Estados miembros y el titular de la autorización de comercialización
del medicamento enviarán al Comité toda la información
disponible sobre la cuestión.
Artículo 32
1. Cuando se recurra al procedimiento descrito en este artículo, el
Comité deliberará y emitirá un dictamen motivado en un
plazo de 90 días a partir de la fecha en que le fue sometida la cuestión.
No obstante, en los casos presentados al Comité con arreglo a los artículos
30 y 31, este plazo podrá prorrogarse 90 días.
En caso de urgencia, a propuesta de su presidente, el Comité podrá
decidir un plazo más corto.
2. Para examinar la cuestión, el Comité podrá designar
como ponente a uno de sus miembros. Podrá también nombrar expertos
independientes para que le asesoren en temas específicos. Al nombrar
a los expertos, el Comité definirá sus funciones y fijará
una fecha límite para la realización de estas funciones.
3. En los casos a que se refieren los artículos 29 y 30, el Comité,
antes de emitir su dictamen, ofrecerá al titular de la autorización
de comercialización la posibilidad de presentar alegaciones orales o
escritas.
En el caso a que se refiere el artículo 31, se podrá solicitar
al titular de la autorización de comercialización que presente
alegaciones orales o escritas.
Si lo considera conveniente, el Comité podrá invitar a cualquier
otra persona a que le proporcione información sobre la materia.
El Comité podrá dejar en suspenso la fecha límite mencionada
en el apartado 1 para que el titular de la autorización de la comercialización
pueda preparar sus alegaciones.
4. La Agencia informará sin demora al titular de la autorización
de comercialización cuando del dictamen del Comité resulte que:
- la solicitud no cumple los criterios de autorización, o
- debe modificarse el resumen de las características del producto propuesto
por el solicitante con arreglo al artículo 11, o
- la autorización debe concederse en determinadas condiciones, teniendo
en cuenta las condiciones que se consideren esenciales para un uso seguro y
eficaz del medicamento incluida la farmacovigilancia, o
- debe suspenderse, modificarse o retirarse una autorización de comercialización.
En un plazo de 15 días a partir de la recepción del dictamen,
el titular podrá notificar por escrito a la Agencia su intención
de recurrir. En tal caso, transmitirá a ésta detalladamente, en
un plazo de 60 días a partir de la recepción del dictamen, los
motivos de su recurso. En un plazo de 60 días a partir de la recepción
de los motivos del recurso, el Comité decidirá si debe revisar
su dictamen y adjuntará al informe de evaluación mencionado en
el apartado 5 las conclusiones sobre el recurso.
5. En un plazo de 30 días a partir de su adopción, la Agencia
presentará el dictamen definitivo del Comité a la Comisión,
a los Estados miembros y al titular de la autorización de comercialización,
junto con un informe en el que se explique la evaluación del medicamento
y se expongan las razones en las que se basan sus conclusiones.
En caso de que el dictamen sea favorable a la concesión de una autorización
de comercialización, se adjuntarán al dictamen los siguientes
documentos:
a) un proyecto de resumen de las características del producto, tal como
se indica en el artículo 11;
b) en su caso, las condiciones en las que la autorización debe concederse
con arreglo al apartado 4.
Artículo 33
En un plazo de 30 días a partir de la recepción del dictamen,
la Comisión preparará un proyecto de decisión respecto
de la solicitud, teniendo en cuenta el Derecho comunitario.
En el caso de un proyecto de decisión que prevea la concesión
de una autorización de comercialización, se adjuntarán
los documentos mencionados en las letras a) y b) del apartado 5 del artículo
32.
En el caso excepcional de que el proyecto de decisión difiera del dictamen
de la Agencia, la Comisión adjuntará asimismo una explicación
detallada de las razones de las diferencias.
El proyecto de decisión se enviará a los Estados miembros y al
solicitante.
Artículo 34
1. La decisión definitiva sobre la solicitud se adoptará con
arreglo al procedimiento establecido en el apartado 2 del artículo 121.
2. Las normas de procedimiento del Comité permanente creado en el apartado
1 del artículo 121 se modificarán para tener en cuenta las tareas
que le corresponden de conformidad con el presente Capítulo.
En dichas modificaciones se incluirá lo siguiente:
- salvo en los casos contemplados en el párrafo tercero del artículo
33, el dictamen del Comité permanente se facilitará por escrito;
- cada Estado miembro contará con 28 días como mínimo para
enviar a la Comisión observaciones por escrito sobre el proyecto de decisión;
- cada Estado miembro podrá solicitar por escrito que el proyecto de
decisión sea debatido por el Comité permanente, pormenorizando
sus motivos.
En el caso de que, a juicio de la Comisión, las observaciones por escrito
de un Estado miembro planteen nuevas cuestiones importantes de índole
científica o técnica que no se hayan abordado en el dictamen de
la Agencia, el presidente interrumpirá el procedimiento y devolverá
la solicitud a la Agencia para un nuevo examen.
La Comisión adoptará las medidas necesarias para la aplicación
de este apartado, de conformidad con el procedimiento previsto en el apartado
2 del artículo 121.
3. Toda decisión adoptada de conformidad con el apartado 1 deberá
enviarse a los Estados miembros a quienes concierna el asunto y ser comunicada
al titular de la autorización de comercialización. Los Estados
miembros autorizarán o retirarán la autorización de comercialización
o introducirán en la autorización las modificaciones que sean
necesarias para ajustarse a la decisión dentro de los 30 días
siguientes a la notificación de ésta. Informarán de ello
a la Comisión y a la Agencia.
Artículo 35
1. Toda solicitud presentada por el titular de la autorización de comercialización
para modificar una autorización de comercialización concedida
según lo dispuesto en el presente capítulo deberá presentarse
a todos los Estados miembros que hubieren autorizado previamente el medicamento
de que se trate.
La Comisión, previa consulta a la Agencia, adoptará las disposiciones
adecuadas para el examen de las modificaciones introducidas en los términos
de una autorización de comercialización.
Estas disposiciones comprenderán un sistema de notificación o
procedimientos administrativos relativos a las modificaciones de importancia
menor y definirán con precisión el concepto de "modificación
de importancia menor".
La Comisión adoptará dichas disposiciones por medio de un reglamento
de aplicación, con arreglo al procedimiento contemplado en el apartado
2 del artículo 121.
2. En caso de arbitraje sometido a la Comisión, los procedimientos establecidos
en los artículos 32, 33 y 34 se aplicarán mutatis mutandis a las
modificaciones de las autorizaciones de comercialización.
Artículo 36
1. Cuando un Estado miembro considere que la modificación de los términos
de una autorización de comercialización concedida según
lo dispuesto en el presente capítulo, o su suspensión o retirada,
son necesarias para proteger la salud pública, someterá sin demora
el asunto a la Agencia para que se apliquen los procedimientos establecidos
en los artículos 32, 33 y 34.
2. Sin perjuicio de las disposiciones del artículo 31, en casos excepcionales,
cuando sea indispensable una acción urgente para proteger la salud pública,
y hasta que se tome una decisión definitiva, el Estado miembro podrá
suspender la comercialización y la utilización en su territorio
del medicamento de que se trate. A más tardar el día hábil
siguiente, dicho Estado informará a la Comisión y a los demás
Estados miembros de los motivos de dicha medida.
Artículo 37
Los artículos 35 y 36 se aplicarán por analogía a los medicamentos autorizados por los Estados miembros previo dictamen del Comité emitido según lo dispuesto en el artículo 4 de la Directiva 87/22/CEE antes del 1 de enero de 1995.
Artículo 38
1. La Agencia publicará un informe anual sobre el funcionamiento de
los procedimientos descritos en el presente capítulo que remitirá
para información al Parlamento Europeo y al Consejo.
2. Antes del 1 de enero de 2001, la Comisión publicará un informe
detallado sobre la aplicación de los procedimientos establecidos en el
presente capítulo y propondrá las modificaciones que sean necesarias
para mejorar dichos procedimientos.
El Consejo, en las condiciones previstas en el Tratado, decidirá sobre
la propuesta de la Comisión en un plazo de un año a partir de
su presentación.
Artículo 39
Las disposiciones de los artículos 27 a 34 no se aplicarán a los medicamentos homeopáticos mencionados en el apartado 2 del artículo 16.
TÍTULO IV
FABRICACIÓN E IMPORTACIÓN
Artículo 40
1. Los Estados miembros tomarán todas las disposiciones útiles
para que la fabricación de medicamentos en su territorio se supedite
a la posesión de una autorización. Esta autorización de
fabricación será necesaria incluso si el medicamento se fabricare
para su exportación.
2. La autorización mencionada en el apartado 1 se exigirá tanto
para la fabricación total o parcial como para las operaciones de división,
de acondicionamiento o de presentación.
No obstante, esta autorización no se exigirá en el caso de las
preparaciones, divisiones y cambios de acondicionamiento o de presentación,
en la medida en que estas operaciones fueran realizadas, únicamente con
vistas a su despacho al por menor, por farmacéuticos en un laboratorio
o por otras personas legalmente autorizadas en los Estados miembros para efectuar
dichas operaciones.
3. La autorización mencionada en el apartado 1 se exigirá igualmente
para las importaciones de medicamentos procedentes de terceros países
en un Estado miembro; con este fin, el presente título y el artículo
118 se aplicarán a tales importaciones de la misma manera en que se aplican
a la fabricación.
Artículo 41
Para obtener la autorización de fabricación, el solicitante deberá
satisfacer, al menos, las exigencias siguientes:
a) especificar los medicamentos y las formas farmacéuticas que se vayan
a fabricar o importar, así como el lugar de su fabricación y/o
de su control;
b) disponer, para su fabricación o importación, de locales, equipo
técnico y posibilidades de control apropiadas y suficientes, que respondan
a las exigencias legales que el Estado miembro interesado prevea, tanto desde
el punto de vista de la fabricación y del control, como de la conservación
de los medicamentos, en la observancia de las disposiciones del artículo
20;
c) disponer al menos de una persona cualificada en el sentido del artículo
48.
El solicitante deberá facilitar en su solicitud los datos justificativos.
Artículo 42
1. La autoridad competente del Estado miembro sólo concederá
la autorización de fabricación después de asegurarse, mediante
una investigación realizada por sus agentes, de que la información
facilitada en virtud del artículo 41 es exacta.
2. La autorización podrá ir acompañada, para garantizar
la observancia de las condiciones previstas en el artículo 41, por determinadas
obligaciones impuestas, bien en el momento de su otorgamiento, bien, posteriormente,
en el de su concesión.
3. La autorización no se aplicará más que a los locales
indicados en la solicitud, así como a los medicamentos y a las formas
farmacéuticas indicadas en dicha solicitud.
Artículo 43
Los Estados miembros adoptarán todas las disposiciones adecuadas para que la duración del procedimiento para el otorgamiento de la autorización de fabricación no exceda un plazo de 90 días, a partir de la fecha de recepción de la solicitud por la autoridad competente.
Artículo 44
En caso de solicitud de modificación por parte del titular de la autorización de fabricación de uno de los elementos contemplados en las letras a) y b) del párrafo primero del artículo 41, la duración del procedimiento relativo a esta demanda no rebasará los 30 días. En casos excepcionales, este plazo podrá ser prorrogado hasta 90 días.
Artículo 45
La autoridad competente del Estado miembro podrá exigir del solicitante información complementaria en lo referente a la información facilitada en aplicación del artículo 41, así como en lo relativo a la persona cualificada contemplada en el artículo 48; cuando la autoridad competente alegara esta facultad, los plazos previstos en los artículos 43 y 44 quedarán interrumpidos hasta que los datos complementarios requeridos hubieran sido facilitados.
Artículo 46
El titular de la autorización de fabricación estará obligado,
al menos:
a) a disponer del personal que responda a las exigencias legales previstas por
el Estado miembro interesado, tanto desde el punto de vista de la fabricación
como de los controles;
b) a no vender los medicamentos autorizados más que de acuerdo con la
legislación de los Estados miembros interesados;
c) a informar previamente a la autoridad competente de toda modificación
que deseara aportar a cualquiera de los informes suministrados en aplicación
del artículo 41; sin embargo, la autoridad competente será informada
sin demora en caso de sustitución imprevista de la persona cualificada
mencionada en el artículo 48;
d) a permitir, en todo momento, el acceso a sus locales a los agentes de la
autoridad competente del Estado miembro interesado;
e) a permitir que la persona cualificada mencionada en el artículo 48
pueda cumplir su misión, en particular poniendo a su disposición
todos los medios necesarios;
f) a respetar los principios y directrices de las prácticas correctas
de fabricación de los productos farmacéuticos previstos en el
Derecho comunitario.
Artículo 47
Los principios y directrices de las prácticas correctas de fabricación
para los medicamentos que se contemplan en la letra f) del artículo 46
se adoptarán en forma de Directiva, de conformidad con el apartado 2
del artículo 121.
La Comisión publicará detalladamente las líneas directrices
con arreglo a dichos principios y los revisará cuando sea necesario para
tener en cuenta los progresos técnicos y científicos.
Artículo 48
1. Los Estados miembros adoptarán todas las disposiciones adecuadas,
para que el titular de la autorización de fabricación disponga
de forma permanente y continua, al menos de una persona cualificada que responda
a las condiciones previstas en el artículo 49, responsable en particular
de la ejecución de las obligaciones especificadas en el artículo
51.
2. En el caso en que respondiese personalmente a las condiciones previstas en
el artículo 49, el titular de la autorización podrá asumir
por sí mismo la responsabilidad mencionada en el apartado 1.
Artículo 49
1. Los Estados miembros velarán para que la persona cualificada mencionada
en el artículo 48 responda a las condiciones mínimas de cualificación
previstas en los apartados 2 y 3.
2. La persona cualificada deberá estar en posesión de un diploma,
certificado u otro título que sancione un ciclo de formación universitaria
-o un ciclo de formación reconocido como equivalente por el Estado miembro
interesado- que tenga una duración mínima de cuatro años
de enseñanza teórica y práctica en una de las especialidades
científicas siguientes: farmacia, medicina, veterinaria, química,
química y tecnología farmacéuticas, biología.
No obstante, la duración mínima del ciclo de formación
universitaria podrá ser de tres años y medio cuando dicho ciclo
esté seguido por un período de formación teórica
y práctica de una duración mínima de un año, y que
incluya un período de prácticas de al menos seis meses en una
farmacia abierta al público, y sancionado por un examen de nivel universitario.
Cuando coexistan en un Estado miembro dos ciclos de formación universitaria
o reconocidos como equivalentes por dicho Estado, uno de los cuales tenga una
duración de cuatro años y el otro de tres años, el diploma,
certificado u otro título que sancione el ciclo de formación universitaria
-o reconocido como equivalente- de tres años se considerará que
cumple los requisitos de duración previstos en el segundo párrafo
siempre que los diplomas, certificados u otros títulos que sancionen
los dos ciclos de formación sean reconocidos como equivalentes por dicho
Estado.
El ciclo de formación contendrá una enseñanza teórica
y práctica que puede versar al menos sobre las asignaturas básicas
siguientes:
- física experimental,
- química general e inorgánica,
- química orgánica,
- química analítica,
- química farmacéutica, incluyendo el análisis de medicamentos,
- bioquímica general y aplicada (médica),
- fisiología,
- microbiología,
- farmacología,
- tecnología farmacéutica,
- toxicología,
- farmacognosia (asignatura médica) (estudio de la composición
y efectos de las sustancias activas naturales de origen vegetal o animal).
La enseñanza de estas asignaturas deberá ser dosificada de manera
que permita al interesado asumir las obligaciones especificadas en el artículo
51.
En la medida en que algunos diplomas, certificados u otros títulos enumerados
en el primer párrafo no respeten los criterios establecidos en el presente
apartado, la autoridad competente del Estado miembro se asegurará de
que el interesado haya dado prueba de conocimientos satisfactorios en el ámbito
de las asignaturas en cuestión.
3. La persona cualificada deberá haber ejercido, durante al menos dos
años y en una o varias empresas que hayan obtenido una autorización
de fabricación, de actividades de análisis cualitativo de medicamentos,
de análisis cuantitativo de sustancias activas, así como de pruebas
y verificaciones necesarias para asegurar la calidad de los medicamentos.
La duración de la experiencia práctica podrá disminuirse
en un año cuando el ciclo de formación universitaria tenga una
duración de al menos cinco años, y en un año y medio cuando
dicho ciclo de formación tenga una duración mínima de seis
años.
Artículo 50
1. Toda persona que ejerza en un Estado miembro las actividades de la persona
contemplada en el artículo 48 en el momento de la puesta en aplicación
de la Directiva 75/319/CEE en dicho Estado, sin ajustarse a las disposiciones
del artículo 49, estará cualificada para continuar ejerciendo
tales actividades en dicho Estado.
2. El titular de un diploma, certificado u otro título, que sancione
un ciclo de formación universitaria u otro ciclo reconocido como equivalente
por el Estado miembro interesado en una disciplina científica que le
habilite para el ejercicio de las actividades de la persona contemplada en el
artículo 48, con arreglo a la legislación de dicho Estado, podrá
cuando haya comenzado su formación antes del 21 de mayo de 1975 considerarse
cualificado para asumir en este Estado las tareas propias de la persona a que
se refiere el artículo 48, a condición de haber ejercido previamente
y antes del 21 de mayo de 1985, durante al menos dos años y, en una o
varias empresas que hubieran obtenido una autorización de fabricación,
actividades de control de producción y/o actividades de análisis
cuantitativo y cualitativo de las sustancias activas, así como pruebas
y verificaciones necesarias para asegurar la calidad de los medicamentos bajo
la autoridad directa de una persona contemplada en el artículo 48.
Cuando el interesado haya adquirido la experiencia práctica a que se
refiere el párrafo primero antes del 21 de mayo de 1965, se le exigirá
un año suplementario de experiencia práctica que responda a las
condiciones del párrafo primero y que haya sido efectuada inmediatamente
antes del ejercicio de estas actividades.
Artículo 51
1. Los Estados miembros adoptarán todas las disposiciones adecuadas
para que la persona cualificada contemplada en artículo 48, sin perjuicio
de sus relaciones con el titular de la autorización de fabricación,
tenga la responsabilidad, en el marco de los procedimientos mencionados en el
artículo 52, de procurar que:
a) en el caso de medicamentos fabricados en el Estado miembro interesado, cada
lote de medicamentos haya sido fabricado y controlado con arreglo a la legislación
en vigor en dicho Estado miembro y en la observancia de las exigencias requeridas
para la autorización de comercialización;
b) en el caso de los medicamentos procedentes de terceros países, cada
lote de fabricación importado haya sido objeto, en el Estado miembro
importador, de un análisis cualitativo completo, un análisis cuantitativo
de al menos todas las sustancias activas y de todas las demás pruebas
o verificaciones necesarias para asegurar la calidad de medicamentos en la observancia
de las exigencias requeridas para la autorización de comercialización.
Los lotes de medicamentos así controlados en un Estado miembro quedarán
exceptuados de los citados controles cuando sean comercializados en otro Estado
miembro, acompañados de las actas de control firmadas por la persona
cualificada.
2. En el caso de medicamentos importados de terceros países, si la Comunidad
hubiere adoptado con el país exportador disposiciones adecuadas que garanticen
que el fabricante del medicamento aplica prácticas correctas de fabricación
por lo menos equivalentes a las establecidas por la Comunidad y que se han efectuado
en el país exportador los controles mencionados en la letra b) del párrafo
primero del apartado 1, la persona encargada de efectuar los controles podrá
ser dispensada de realizarlos.
3. En todos los casos, y en particular cuando los medicamentos sean expuestos
a la venta, la persona cualificada deberá certificar que cada lote de
fabricación responde a las disposiciones del presente artículo,
en un registro o documento equivalente previsto a este respecto; dicho registro
o documento equivalente deberá tenerse al día a medida que se
vayan efectuando las operaciones, y ponerse a disposición de los agentes
de la autoridad competente durante un período que respete las disposiciones
del Estado miembro interesado y en todo caso durante un período de cinco
años como mínimo.
Artículo 52
Los Estados miembros garantizarán la observancia de las obligaciones
de la persona cualificada a que se refiere el artículo 48, mediante medidas
administrativas apropiadas o mediante el establecimiento de una disciplina profesional.
Los Estados miembros podrán proceder a la suspensión temporal
de dicha persona a partir del momento de la incoación de un procedimiento
administrativo o disciplinario seguido contra ella por incumplimiento de sus
obligaciones.
Artículo 53
Las disposiciones del presente título se aplicarán igualmente a los medicamentos homeopáticos.
TÍTULO V
ETIQUETADO Y PROSPECTO
Artículo 54
El embalaje exterior o, a falta de éste, el acondicionamiento primario
de todo medicamento deberá llevar las indicaciones siguientes:
a) la denominación del medicamento, seguida de la denominación
común cuando el medicamento no contenga más que una única
sustancia activa y su denominación sea un nombre arbitrario; en caso
de existir varias presentaciones y/o varias dosificaciones del mismo medicamento,
en la denominación del medicamento deberá figurar la forma farmacéutica
y/o la dosificación (en caso necesario, lactantes, niños, adultos);
b) la composición cualitativa y cuantitativa en sustancias activas por
unidad de toma o, según la forma de administración para un volumen
o peso determinado, utilizando las denominaciones comunes;
c) la forma farmacéutica y contenido en peso, volumen o unidades de toma;
d) la lista de los excipientes que tengan acción o efecto conocidos y
que estén previstos en las directrices publicadas con arreglo al artículo
65. No obstante, deberán indicarse todos los excipientes cuando se trate
de un producto inyectable, de una preparación tópica o de un colirio;
e) la forma de administración y, si fuere necesario, la vía de
administración;
f) una advertencia especial que indique que el medicamento debe mantenerse fuera
del alcance de los niños;
g) una advertencia especial, cuando el medicamento la requiera;
h) la fecha de caducidad expresada claramente (mes/año);
i) las precauciones particulares de conservación, en su caso;
j) las precauciones especiales de eliminación de los medicamentos no
utilizados o de los residuos derivados de estos medicamentos, en su caso;
k) el nombre y dirección del titular de la autorización de comercialización;
l) el número de autorización de comercialización;
m) el número del lote de fabricación;
n) para los medicamentos de automedicación, la indicación de uso.
Artículo 55
1. Los acondicionamientos primarios distintos de los que se mencionan en los
apartados 2 y 3 deberán llevar las indicaciones previstas en los artículos
54 y 62.
2. Los acondicionamientos primarios que se presenten en forma de blister, cuando
estén contenidos en un embalaje exterior con arreglo a lo dispuesto en
los artículos 54 y 62, deberán llevar, como mínimo, las
indicaciones siguientes:
- la denominación del medicamento tal como se contempla en la letra a)
del artículo 54,
- el nombre del titular de la autorización de comercialización,
- la fecha de caducidad,
- el número del lote de fabricación.
3. Los pequeños acondicionamientos primarios en los que sea imposible
mencionar las informaciones previstas en los artículos 54 y 62 deberán
llevar, como mínimo, las indicaciones siguientes:
- la denominación del medicamento y, si fuere necesario, dosis, vía
de administración,
- la forma de administración,
- la fecha de caducidad,
- el número del lote de fabricación,
- el contenido en peso, en volumen o en unidades.
Artículo 56
Las indicaciones previstas en los artículos 54, 55 y 62 deberán ser fácilmente legibles, claramente comprensibles e indelebles.
Artículo 57
No obstante lo dispuesto en el artículo 60, los Estados miembros podrán
exigir la utilización de determinadas modalidades de etiquetado del medicamento
que permitan indicar:
- la identificación del precio del medicamento,
- la identificación de las condiciones de reembolso por los organismos
de seguridad social,
- la identificación del régimen jurídico con arreglo al
cual el medicamento sea dispensado al paciente, de conformidad con título
VI,
- la identificación y autenticidad.
Artículo 58
La inclusión de un prospecto en el envase de todo medicamento será obligatorio salvo si toda la información exigida en los artículos 59 y 62 figura directamente en el embalaje exterior o en el acondicionamiento primario.
Artículo 59
1. El prospecto se elaborará de conformidad con el resumen de las características
del producto, y deberá incluir los siguientes datos, en este orden:
a) para la identificación del medicamento:
- la denominación del medicamento, seguida de la denominación
común cuando el medicamento no contenga más que una sola sustancia
activa y su denominación sea un nombre arbitrario; cuando para un medicamento
existan varias presentaciones farmacéuticas y/o dosificaciones, la presentación
farmacéutica y/o la dosificación (en caso necesario, lactantes,
niños, adultos) deberán figurar en la denominación del
medicamento;
- la composición cualitativa completa (en sustancias activas y excipientes),
así como la composición cuantitativa en sustancias activas, utilizando
las denominaciones comunes, para cada presentación del medicamento;
- la forma farmacéutica y el contenido en peso, en volumen, o en unidad
de toma, para cada presentación del medicamento;
- la categoría farmacoterapéutica, o el tipo de actividad, en
términos fácilmente comprensibles para el usuario;
- el nombre y la dirección del titular de la autorización de comercialización
y del fabricante;
b) las indicaciones terapéuticas;
c) enumeración de las informaciones necesarias previas a la toma del
medicamento:
- contraindicaciones;
- precauciones de empleo adecuadas;
- interacciones medicamentosas y otras interacciones (por ejemplo: alcohol,
tabaco, alimentos) que puedan afectar a la acción del medicamento;
- advertencias especiales.
Esta enumeración deberá:
- tener en cuenta la situación particular de ciertas categorías
de usuarios (niños, mujeres embarazadas o durante la lactancia, ancianos,
personas con ciertas patologías específicas);
- mencionar, en su caso, los posibles efectos del tratamiento sobre la capacidad
para conducir un vehículo o manipular determinadas máquinas;
- incluir una lista de excipientes cuyo conocimiento sea importante para una
utilización eficaz y sin riesgos del medicamento y que esté establecida
por las directrices publicadas con arreglo a lo dispuesto en el artículo
65;
d) las instrucciones necesarias y habituales para una buena utilización,
en particular:
- la posología;
- la forma y, si fuere necesario, vía de administración;
- la frecuencia de administración, precisando, si fuere necesario, el
momento en que deba o pueda administrarse el medicamento;
y, en su caso, según la naturaleza del producto:
- la duración del tratamiento, cuando tenga que ser limitada;
- las medidas que deban tomarse en caso de sobredosis (por ejemplo: síntomas,
tratamiento de urgencia);
- la actitud que deba tomarse en caso de que se haya omitido la administración
de una o varias dosis;
- la indicación, si es necesario, del riesgo de síndrome de abstinencia;
e) descripción de reacciones adversas que puedan observarse durante el
uso normal del medicamento, y, en su caso, medidas que deban adoptarse; el usuario
será invitado expresamente a comunicar a su médico o a su farmacéutico
cualquier reacción no querida que no estuviere descrita en el prospecto;
f) referencia a la fecha de caducidad que figure en el envase, con:
- una advertencia para no sobrepasar esta fecha;
- si procediere, las precauciones especiales de conservación;
- en su caso, una advertencia con respecto a ciertos signos visibles de deterioro;
g) la fecha de la última revisión del prospecto.
2. No obstante lo dispuesto en la letra b) del apartado 1, la autoridad competente
podrá decidir que ciertas indicaciones terapéuticas no figuren
en el prospecto, cuando la difusión de esta información pueda
implicar graves inconvenientes para el paciente.
Artículo 60
Los Estados miembros no podrán prohibir ni impedir la comercialización de medicamentos en su territorio por motivos relacionados con el etiquetado o el prospecto si éstos se ajustan a las disposiciones del presente título.
Artículo 61
1. Al solicitar la autorización para la comercialización, se
presentará a las autoridades competentes en materia de autorización
de comercialización, una o varias muestras o maquetas del embalaje exterior
y del acondicionamiento primario, así como el proyecto de prospecto.
2. La autoridad competente no se opondrá a la comercialización
del medicamento si el etiquetado o el prospecto cumplen lo dispuesto en el presente
título, y son conformes con la información que figura en el resumen
de las características del producto.
3. Cualquier proyecto de modificación de un elemento relativo al etiquetado
o al prospecto regulado por el presente título que no esté relacionada
con el resumen de las características del producto, será presentado
a las autoridades competentes en materia de autorización de comercialización.
Si las autoridades competentes no se pronuncian contra el proyecto de modificación
en el plazo de 90 días a partir de la fecha de presentación de
la solicitud, el solicitante podrá proceder a la realización de
las modificaciones.
4. La circunstancia de que las autoridades competentes no se hayan opuesto a
la comercialización del medicamento en aplicación del apartado
2 o a una modificación del etiquetado o del prospecto en aplicación
del apartado 3, no afectará a la responsabilidad de Derecho común
del fabricante ni, en su caso, del titular de la autorización de comercialización.
Artículo 62
El embalaje exterior y el prospecto pueden llevar signos o dibujos tendentes a explicar determinadas informaciones contempladas en los artículos 54 y 59 así como otras informaciones compatibles con el resumen de las características del producto, útiles para la educación sanitaria, con exclusión de cualquier elemento que pueda tener carácter publicitario.
Artículo 63
1. Las indicaciones previstas en los artículos 54, 59 y 62 para el etiquetado
deberán redactarse en la lengua o lenguas oficiales del Estado miembro
de comercialización.
La disposición del párrafo primero no será obstáculo
para que dichas indicaciones estén redactadas en varias lenguas, siempre
que en todas las lenguas utilizadas figuren las mismas indicaciones.
2. El prospecto deberá estar redactado en términos claros y comprensibles
para los usuarios, en la lengua o lenguas oficiales del Estado miembro de comercialización
y de manera que resulte fácilmente legible.
La disposición del párrafo primero no será obstáculo
para que el prospecto esté redactado en varias lenguas, siempre que en
todas las lenguas utilizadas figure la misma información.
3. Cuando el destino del medicamento no sea el suministro al paciente con fines
de automedicación, las autoridades competentes podrán dispensar
de la obligación de hacer figurar determinadas indicaciones en las etiquetas
y en los prospectos de medicamentos específicos y de redactar el prospecto
en la lengua o lenguas oficiales del Estado miembro de comercialización.
Artículo 64
En caso de incumplimiento de lo dispuesto en el presente título, las autoridades competentes de los Estados miembros, si sus requerimientos al interesado no dan resultado, podrán proceder a la suspensión de la autorización de comercialización, hasta que el etiquetado y el prospecto del medicamento de que se trate se ajusten a las disposiciones del presente título.
Artículo 65
De ser necesario, la Comisión publicará directrices especialmente
sobre:
- la formulación de ciertas advertencias especiales para determinadas
categorías de medicamentos;
- las necesidades particulares de información relativas a la automedicación;
- la legibilidad de las indicaciones que figuren en el etiquetado y en el prospecto;
- los métodos de identificación y de autentificación de
los medicamentos;
- la lista de los excipientes que deberán figurar en el etiquetado de
los medicamentos, así como la forma en que deben indicarse dichos excipientes.
Dichas directrices adoptarán la forma de una Directiva, con arreglo al
procedimiento previsto en el apartado 2 del artículo 121.
Artículo 66
1. El embalaje exterior y el envase de los medicamentos que contengan radionucleidos
estarán etiquetados con arreglo a las disposiciones para el transporte
seguro de materiales radiactivos establecidas por el Organismo Internacional
de la Energía Atómica. Además, el etiquetado se ajustará
a las siguientes disposiciones previstas en los apartados 2 y 3.
2. El etiquetado del blindaje de protección incluirá las informaciones
mencionadas en el artículo 54. Además, el etiquetado del blindaje
de protección explicará completamente los códigos utilizados
en el vial e indicará, en su caso, para un tiempo y fecha dados, la cantidad
de radiactividad por dosis o por vial y el número de cápsulas
o, si se trata de líquidos, el número de mililitros contenidos
en el envase.
3. El vial irá etiquetado con la siguiente información:
- el nombre o código del medicamento, con inclusión del nombre
o símbolo químico del radionucleido,
- la indicación del lote y fecha de caducidad,
- el símbolo internacional de radiactividad,
- el nombre del fabricante,
- la cantidad de radiactividad según se indica en el apartado 2.
Artículo 67
La autoridad competente verificará que se adjunte un prospecto informativo detallado en el envase de los radiofármacos, generadores de radionucleidos, equipos reactivos de radionucleidos o precursores de radionucleidos. El texto de dicho prospecto se redactará de acuerdo con lo dispuesto en el artículo 59. Además, el prospecto incluirá todas las precauciones que deban tomar el usuario y el paciente durante la preparación y administración del medicamento, así como las precauciones especiales que deban adoptarse para la eliminación del envase y de su contenido no utilizado.
Artículo 68
Sin perjuicio de las disposiciones del artículo 69, los medicamentos homeopáticos deberán estar etiquetados de conformidad con las disposiciones del presente título e identificarse mediante la mención de su naturaleza homeopática, en caracteres claros y legibles.
Artículo 69
1. En el etiquetado y, en su caso, en el prospecto de los medicamentos contemplados
en el apartado 1 del artículo 14, además de la indicación
"medicamento homeopático" bien visible, constarán única
y obligatoriamente los siguientes datos:
- la denominación científica de la cepa o cepas, seguida del grado
de dilución, empleando los símbolos de la farmacopea utilizada
de conformidad con el punto 5 del artículo 1;
- el nombre y dirección del titular de la autorización de comercialización
y, en su caso, del fabricante;
- el modo de administración y, si fuere necesario, la vía de administración;
- la fecha de caducidad en forma clara (mes y año);
- la fórmula galénica;
- el contenido del modelo de venta;
- las precauciones específicas de conservación, cuando proceda;
- advertencias especiales si el medicamento así lo exige;
- el número de lote de fabricación;
- el número de registro;
- medicamento homeopático "sin indicaciones terapéuticas
aprobadas";
- una advertencia que aconseje al utilizador que consulte a un médico
si los síntomas persisten durante la utilización del medicamento.
2. No obstante lo dispuesto en el apartado 1, los Estados miembros podrán
exigir la utilización de determinadas modalidades de etiquetado que permitan
la indicación:
- del precio del medicamento;
- de las condiciones de reembolso por los organismos de la seguridad social.
TÍTULO VI
CLASIFICACIÓN DE LOS MEDICAMENTOS
Artículo 70
1. Al autorizar la comercialización de un medicamento, las autoridades
competentes especificarán la clasificación del mismo como:
- medicamento sujeto a receta médica,
- medicamento no sujeto a receta médica.
Con tal fin aplicarán los criterios enumerados en el apartado 1 del artículo
71.
2. Las autoridades competentes podrán establecer subcategorías,
en lo que se refiere a los medicamentos que sólo pueden dispensarse con
receta médica. En tal caso, se referirán a la clasificación
siguiente:
a) medicamentos con receta médica renovable o no renovable,
b) medicamentos sujetos a receta médica especial,
c) medicamentos con receta médica restringida, reservados a determinados
medios especializados.
Artículo 71
1. Los medicamentos estarán sujetos a receta médica cuando:
- puedan presentar un peligro, directa o indirectamente, incluso en condiciones
normales de uso, si se utilizan sin control médico, o
- se utilicen frecuentemente, y de forma muy considerable, en condiciones anormales
de utilización, y ello pueda suponer, directa o indirectamente, un peligro
para la salud, o
- contengan sustancias o preparados a base de dichas sustancias, cuya actividad
y/o reacciones adversas sea necesario estudiar más detalladamente, o
- se administren por vía parenteral, salvo casos excepcionales, por prescripción
médica.
2. Cuando los Estados miembros establezcan la subcategoría de los medicamentos
sujetos a receta médica especial, tendrán en cuenta los siguientes
elementos:
- que el medicamento contenga, en una dosis no exenta, una sustancia clasificada
como estupefaciente o psicotropo con arreglo a los convenios internacionales
como el Convenio de las Naciones Unidas de 1961 y 1971, o
- que el medicamento pueda ser objeto, en caso de utilización anormal,
de riesgo considerable de abuso medicamentoso, pueda provocar tóxicodependencia
o ser desviado para usos ilegales, o
- que el medicamento contenga una sustancia que, por su novedad o propiedades,
pudiera considerarse como perteneciente al grupo contemplado en el segundo guión,
como medida de precaución.
3. Cuando los Estados miembros establezcan la subcategoría de los medicamentos
sujetos a receta médica restringida, tendrán en cuenta los siguientes
elementos:
- que el medicamento, a causa de sus características farmacológicas
o por su novedad, o por motivos de salud pública, se reserve para tratamientos
que sólo pueden seguirse en medio hospitalario,
- que el medicamento se utilice en el tratamiento de enfermedades que deban
ser diagnosticadas en medio hospitalario, o en establecimientos que dispongan
de medios de diagnóstico adecuados, aunque la administración y
seguimiento pueda realizarse fuera del hospital, o
- que el medicamento esté destinado a pacientes ambulatorios, pero cuya
utilización pueda producir reacciones adversas muy graves, lo cual requiere,
si es preciso, una receta médica extendida por un especialista y una
vigilancia especial durante el tratamiento.
4. Las autoridades competentes podrán establecer excepciones a la aplicación
de los apartados 1, 2 y 3 teniendo en cuenta:
a) la dosis máxima única o la dosis máxima diaria, la dosificación,
la forma farmacéutica, determinados envases y/o
b) otras condiciones de utilización que dichas autoridades hayan determinado.
5. Cuando una autoridad competente no clasifique un medicamento en una de las
subcategorías indicadas en el apartado 2 del artículo 70, deberá,
no obstante, tener en cuenta los criterios contemplados en los apartados 2 y
3 del presente artículo para determinar si se debe clasificar un medicamento
en la categoría de los medicamentos que sólo pueden dispensarse
con receta médica.
Artículo 72
Los medicamentos no sujetos a receta médica serán los que no respondan a los criterios expuestos en el artículo 71.
Artículo 73
Las autoridades competentes establecerán la lista de los medicamentos que en su territorio sólo puedan dispensarse con receta médica, con indicación, si fuere necesario, de la categoría de clasificación. Actualizarán dicha lista cada año.
Artículo 74
Con ocasión de la renovación quinquenal de la autorización de comercialización, o cuando se pongan en conocimiento de las autoridades competentes nuevos elementos científicos, dichas autoridades reexaminarán y, en su caso, modificarán la clasificación de un medicamento, aplicando los criterios establecidos en el artículo 71.
Artículo 75
Cada año, los Estados miembros comunicarán a la Comisión y a los demás Estados miembros las modificaciones que hayan introducido en la lista citada en el artículo 73.
TÍTULO VII
DISTRIBUCIÓN AL POR MAYOR DE MEDICAMENTOS
Artículo 76
Sin perjuicio de lo dispuesto en el artículo 6, los Estados miembros adoptarán todas las medidas adecuadas para que en su territorio sólo se distribuyan medicamentos cubiertos por una autorización de comercialización concedida con arreglo al Derecho comunitario.
Artículo 77
1. Los Estados miembros adoptarán todas las disposiciones adecuadas
para que la distribución al por mayor de medicamentos esté sujeta
a la posesión de una autorización para ejercer la actividad de
mayorista de medicamentos en la que se especifique el lugar para el que es válida.
2. Cuando las personas autorizadas o facultadas para dispensar medicamentos
al público puedan asimismo, con arreglo a su legislación nacional,
ejercer una actividad al por mayor, dichas personas estarán sometidas
a la autorización establecida en el apartado 1.
3. La posesión de una autorización de fabricación implica
la de distribuir al por mayor los medicamentos a que se refiere dicha autorización.
La posesión de una autorización para ejercer la actividad de mayorista
de medicamentos no dispensará de la obligación de poseer la autorización
de fabricación y respetar las condiciones establecidas a este respecto,
ni siquiera cuando la actividad de fabricación o de importación
se ejerza de forma accesoria.
4. Cuando lo solicite la Comisión o un Estado miembro, los Estados miembros
estarán obligados a suministrar toda la información pertinente
relativa a las autorizaciones individuales que hayan concedido en virtud del
apartado 1.
5. El control de las personas autorizadas para ejercer la actividad de mayoristas
de medicamentos y la inspección de los locales de que dispongan, serán
efectuados bajo la responsabilidad del Estado miembro que haya concedido la
autorización.
6. El Estado miembro que haya concedido la autorización contemplada en
el apartado 1 suspenderá o retirará dicha autorización
si dejaran de cumplirse las condiciones de autorización. Informará
inmediatamente de ello a los demás Estados miembros y a la Comisión.
7. Si un Estado miembro estimare que no se respetan o han dejado de respetarse
las condiciones de autorización en lo relativo al titular de una autorización
concedida por otro Estado miembro en virtud del apartado 1, informará
inmediatamente de ello a la Comisión y al otro Estado miembro interesado.
Éste adoptará todas las medidas necesarias y comunicará
a la Comisión y al primer Estado miembro las decisiones adoptadas y los
motivos de éstas.
Artículo 78
Los Estados miembros velarán para que la duración del procedimiento
para el estudio de la solicitud de la autorización de distribución
no exceda de 90 días a partir de la fecha de recepción de la solicitud
por la autoridad competente del Estado miembro de que se trate.
En su caso, la autoridad competente podrá exigir al solicitante que proporcione
todas las informaciones necesarias relativas a las condiciones de autorización.
Cuando la autoridad competente haga uso de esa facultad, el plazo que establece
el primer párrafo quedará en suspenso hasta que se hayan proporcionado
los datos complementarios requeridos.
Artículo 79
Para obtener la autorización de distribución, el solicitante
deberá cumplir al menos los requisitos siguientes:
a) disponer de locales, instalaciones y equipos adaptados y suficientes, de
forma que queden garantizadas la buena conservación y buena distribución
de los medicamentos;
b) disponer de personal y en particular de una persona designada como responsable,
cualificada en las condiciones establecidas por la legislación del Estado
miembro de que se trate;
c) comprometerse a cumplir las obligaciones que le correspondan en virtud del
artículo 80.
Artículo 80
El titular de una autorización de distribución estará
obligado a cumplir al menos los requisitos siguientes:
a) facilitar en cualquier momento a los agentes encargados de su inspección
el acceso a los locales, instalaciones y equipos a que se refiere la letra a)
del artículo 79;
b) obtener sus suministros de medicamentos solamente de las personas que posean
la autorización de distribución o que estén dispensadas
de dicha autorización en virtud del apartado 3 del artículo 77;
c) proporcionar medicamentos sólo a personas que posean la autorización
de distribución o que estén autorizadas o facultadas en el Estado
miembro de que se trate para dispensar medicamentos al público;
d) disponer de un plan de emergencia que garantice la aplicación efectiva
de cualquier retirada del mercado ordenada por las autoridades competentes o
iniciada en cooperación con el fabricante del medicamento de que se trate
o el titular de la autorización de comercialización para dicho
medicamento;
e) conservar una documentación, en forma de facturas de compras y ventas,
en forma informatizada o de cualquier otra forma, que incluya al menos los datos
siguientes de toda transacción de entrada y de salida:
- fecha,
- denominación del medicamento,
- cantidad recibida o suministrada,
- nombre y dirección del proveedor o del destinatario, según proceda;
f) tener a disposición de las autoridades competentes, con fines de inspección,
durante un período de cinco años, la documentación contemplada
en la letra e);
g) respetar los principios y directrices de las prácticas correctas de
distribución establecidas en el artículo 84.
Artículo 81
En lo que respecta al suministro de medicamentos a los farmacéuticos
y personas autorizadas o facultadas para dispensar medicamentos al público,
los Estados miembros no impondrán al titular de una autorización
de distribución, concedida por otro Estado miembro, ninguna obligación,
en especial de servicio público, más estricta que las que impongan
a las personas a las que ellos mismos hayan autorizado para ejercer una actividad
equivalente.
Además, es conveniente que estas obligaciones estén justificadas,
de conformidad con el Tratado, por razones de protección de la salud
pública y proporcionada al objetivo relativo a dicha protección.
Artículo 82
Para cualquier suministro de medicamentos a una persona autorizada o facultada
para dispensar medicamentos al público en el Estado miembro de que se
trate, el mayorista autorizado deberá adjuntar todo documento que permita
conocer:
- la fecha,
- la denominación y la forma farmacéutica del medicamento,
- la cantidad suministrada,
- el nombre y la dirección del proveedor y del destinatario.
Los Estados miembros adoptarán las medidas adecuadas para garantizar
que las personas autorizadas o facultadas para dispensar medicamentos al público
faciliten las informaciones que permitan conocer la vía de distribución
de cada medicamento.
Artículo 83
Las disposiciones del presente título se entienden sin perjuicio de
los requisitos más estrictos que los Estados miembros exijan para la
distribución al por mayor de:
- las sustancias narcóticas o psicotrópicas en su territorio;
- los medicamentos derivados de la sangre;
- los medicamentos inmunológicos;
- los medicamentos radiofármacos.
Artículo 84
La Comisión publicará líneas directrices sobre las prácticas correctas de distribución. Consultará a este respecto al Comité de especialidades farmacéuticas y al Comité farmacéutico establecido por la Decisión 75/320/CEE del Consejo(22).
Artículo 85
Las disposiciones del presente título serán aplicables a los medicamentos homeopáticos, con excepción de los contemplados en el apartado 1 del artículo 14.
TÍTULO VIII
PUBLICIDAD
Artículo 86
1. A efectos del presente título, se entenderá por "publicidad
de medicamentos" toda forma de oferta informativa, de prospección
o de incitación destinada a promover la prescripción, la dispensación,
la venta o el consumo de medicamentos; comprenderá en particular:
- la publicidad de medicamentos destinada al público;
- la publicidad de medicamentos destinada a personas facultadas para prescribirlos
o dispensarlos;
- la visita de los visitadores médicos a personas facultadas para prescribir
o dispensar medicamentos;
- el suministro de muestras;
- la incitación a prescribir o dispensar medicamentos mediante concesión,
oferta o promesa de ventajas, pecuniarias o en especie, excepto cuando su valor
intrínseco resulte mínimo;
- el patrocinio de reuniones promocionales a las que asistan personas facultadas
para prescribir o dispensar medicamentos;
- el patrocinio de congresos científicos en los que participen personas
facultadas para prescribir o dispensar medicamentos y, en particular, el hecho
de correr a cargo con los gastos de desplazamiento y estancia con motivo de
dichos congresos.
2. El presente título no contempla:
- el etiquetado y el prospecto, sujetos a las disposiciones del Título
V;
- la correspondencia, acompañada, en su caso, de cualquier documento
no publicitario, necesaria para responder a una pregunta concreta sobre un medicamento
en particular;
- las informaciones concretas y los documentos de referencia relativos, por
ejemplo, al cambio de envase, a las advertencias relativas a reacciones adversas
en el marco de la farmacovigilancia, a los catálogos de ventas y a las
listas de precios siempre que no figure ninguna información sobre el
medicamento;
- la información relativa a la salud humana o a enfermedades de las personas,
siempre que no se haga referencia alguna, ni siquiera indirecta, a un medicamento.
Artículo 87
1. Los Estados miembros prohibirán toda publicidad de un medicamento
para el que no se haya otorgado una autorización de comercialización
de conformidad con el Derecho comunitario.
2. Todos los elementos de la publicidad de un medicamento deberán ajustarse
a las informaciones que figuren en el resumen de las características
del producto.
3. La publicidad referente a un medicamento:
- deberá favorecer la utilización racional del mismo, presentándolo
de forma objetiva y sin exagerar sus propiedades;
- no podrá ser engañosa.
Artículo 88
1. Los Estados miembros prohibirán la publicidad destinada al público
de los medicamentos:
- que sólo pueden dispensarse por prescripción facultativa, con
arreglo al Título VI;
- que contengan sustancias psicotrópicas o estupefacientes, con arreglo
a lo definido en los convenios internacionales, como el Convenio de las Naciones
Unidas de 1961 y 1971;
- que no puedan ser objeto de publicidad destinada al público de acuerdo
con lo dispuesto en el párrafo segundo del apartado 2.
2. Podrán ser objeto de publicidad destinada al público los medicamentos,
que por su composición y objetivo, estén destinados y concebidos
para su utilización sin la intervención de un médico que
realice el diagnóstico, la prescripción o el seguimiento del tratamiento,
en caso necesario tras consultar con el farmacéutico.
Los Estados miembros prohibirán la mención, en la publicidad destinada
al público, de indicaciones terapéuticas tales como:
- la tuberculosis,
- las enfermedades de transmisión sexual,
- otras enfermedades infecciosas graves,
- el cáncer y otras enfermedades tumorales,
- el insomnio crónico,
- la diabetes y otras enfermedades del metabolismo.
3. Los Estados miembros podrán prohibir en su territorio la publicidad
destinada al público de los medicamentos reembolsables.
4. La prohibición establecida en el apartado 1 no será aplicable
a las campañas de vacunación realizadas por la industria y aprobadas
por las autoridades competentes de los Estados miembros.
5. La prohibición establecida en el apartado 1 se aplicará sin
perjuicio de lo dispuesto en el artículo 14 de la Directiva 89/552/CEE.
6. Los Estados miembros prohibirán la distribución directa de
medicamentos al público cuando ésta se realice con fines de promoción
por parte de la industria; no obstante, los Estados miembros podrán autorizar
esta distribución en casos excepcionales y con otros fines.
Artículo 89
1. Sin perjuicio de lo dispuesto en el artículo 88, cualquier tipo de
publicidad de un medicamento que vaya destinada al público deberá:
a) realizarse de manera tal que resulte evidente el carácter publicitario
del mensaje y quede claramente especificado que el producto es un medicamento;
b) incluir como mínimo:
- la denominación del medicamento, así como la denominación
común cuando el medicamento contenga una única sustancia activa,
- las informaciones indispensables para la utilización correcta del medicamento,
- una invitación expresa y claramente visible a leer detenidamente las
instrucciones que figurarán en el prospecto o en el embalaje externo,
según el caso.
2. No obstante lo dispuesto en el apartado 1, los Estados miembros podrán
disponer que la publicidad de un medicamento destinada al público incluya
solamente la denominación del mismo, cuando su único objetivo
sea el de recordar dicha denominación.
Artículo 90
La publicidad de un medicamento destinada al público no podrá
incluir ningún elemento que:
a) atribuya a la consulta médica o a la intervención quirúrgica
un carácter superfluo, especialmente ofreciendo un diagnóstico
o aconsejando un tratamiento por correspondencia;
b) sugiera que el efecto del medicamento está asegurado, que carece de
reacciones adversas o que es superior o igual al de otro tratamiento u otro
medicamento;
c) sugiera que el usuario puede mejorar su salud mediante la utilización
del medicamento;
d) sugiera que la salud del usuario puede verse afectada en caso de no utilización
del medicamento; esta prohibición no se aplicará a las campañas
de vacunación contempladas en el apartado 4 del artículo 88;
e) se dirija, exclusiva o principalmente, a niños;
f) se refiera a una recomendación que hayan formulado científicos,
profesionales de la salud o personas que, aunque no sean científicos
ni profesionales de la salud, puedan, debido a su notoriedad, incitar al consumo
de medicamentos;
g) equipare el medicamento a un producto alimenticio, a un producto cosmético
o a cualquier otro producto de consumo;
h) sugiera que la seguridad o la eficacia del medicamento se debe a que se trata
de una sustancia natural;
i) pueda inducir, mediante una descripción o representación detallada
de la anamnesis, a un falso autodiagnóstico;
j) se refiera de forma abusiva, alarmante o engañosa a testimonios de
curación;
k) utilice de forma abusiva, alarmante o engañosa, representaciones visuales
de las alteraciones del cuerpo humano producidas por enfermedades o lesiones,
o de la acción de un medicamento en el cuerpo humano o en partes del
mismo;
l) mencione que el medicamento ha recibido una autorización de comercialización.
Artículo 91
1. Toda publicidad de un medicamento destinada a personas facultadas para prescribirlo
o dispensarlo deberá incluir:
- las informaciones esenciales compatibles con el resumen de las características
del producto,
- la clasificación del medicamento en materia de dispensación.
Los Estados miembros podrán exigir además que la publicidad incluya
el precio de venta o una tarifa indicativa de las distintas presentaciones y
las condiciones de reembolso por parte de los organismos de seguridad social.
2. Los Estados miembros podrán establecer que la publicidad de un medicamento
destinada a personas facultadas para prescribirlo o dispensarlo pueda, sin perjuicio
de lo dispuesto en el apartado 1, incluir solamente la denominación del
medicamento, cuando su único objetivo sea el de recordar dicha denominación.
Artículo 92
1. Toda documentación relativa a un medicamento que se comunique en
el marco de su promoción ante las personas facultadas para prescribirlo
o dispensarlo, deberá incluir al menos las informaciones contempladas
en el apartado 1 del artículo 91 y precisar la fecha en la que dicha
documentación se haya elaborado o revisado por última vez.
2. Todas las informaciones contenidas en la documentación contemplada
en el apartado 1 deberán ser exactas, actuales, comprobables y lo suficientemente
completas como para permitir que el destinatario se haga una idea propia del
valor terapéutico del medicamento.
3. Las citas, cuadros y otras ilustraciones que se extraigan de revistas médicas
o de obras científicas y que se utilicen en la documentación contemplada
en el apartado 1 deberán reproducirse fielmente, precisando con exactitud
su fuente.
Artículo 93
1. Los visitadores médicos deberán ser formados de manera adecuada
por la empresa que les emplee y poseer conocimientos científicos suficientes
para dar indicaciones precisas y lo más completas posible sobre los medicamentos
que presenten.
2. En cada visita, los visitadores médicos deberán proporcionar
a la persona visitada o tener a su disposición, para cada medicamento
que presenten, el resumen de las características del producto, complementado,
si lo permite la legislación del Estado miembro, con las informaciones
sobre el precio y las condiciones de reembolso citadas en el apartado 1 del
artículo 91.
3. Los visitadores médicos deberán notificar al servicio científico
citado en el apartado 1 del artículo 98 todas las informaciones relativas
a la utilización de los medicamentos de cuya promoción se ocupen,
indicando especialmente las reacciones adversas que las personas visitadas les
comuniquen.
Artículo 94
1. Queda prohibido otorgar, ofrecer o prometer a las personas facultadas para
prescribir o dispensar medicamentos y en el marco de la promoción de
los mismos frente a dichas personas, primas, ventajas pecuniarias o ventajas
en especie, con excepción de aquellas que tengan un valor insignificante
y que sean irrelevantes para la práctica de la medicina o la farmacia.
2. La hospitalidad ofrecida en el marco de manifestaciones de promoción
de los medicamentos deberá ser siempre moderada en su nivel y subordinada
al objetivo principal de la reunión; no podrá ser extensible a
personas que no sean profesionales de la salud.
3. Las personas facultadas para prescribir o dispensar medicamentos no podrán
solicitar o aceptar ninguno de los incentivos prohibidos en virtud del apartado
1 o contrarios a lo dispuesto en el apartado 2.
4. Las medidas o las prácticas comerciales existentes en los Estados
miembros en materia de precios, de márgenes y de descuentos no se verán
afectadas por los apartados 1, 2 y 3.
Artículo 95
Las disposiciones del apartado 1 del artículo 94 no supondrán un obstáculo para la hospitalidad ofrecida, directa o indirectamente, en el marco de manifestaciones de carácter exclusivamente profesional y científico; dicha hospitalidad deberá ser siempre moderada en su nivel y subordinada al objetivo principal de la reunión; no podrá ser extensible a personas que no sean profesionales de la salud.
Artículo 96
1. En casos excepcionales podrán ofrecerse muestras gratuitas exclusivamente
a las personas facultadas para prescribir, y se hará en las condiciones
siguientes:
a) un número limitado de muestras para cada medicamento por año
y persona facultada para la prescripción;
b) cada suministro de muestras deberá responder a una petición
formulada por escrito, fechada y firmada, que proceda del prescriptor;
c) los que suministren las muestras deberán mantener un sistema adecuado
de control y de responsabilidad;
d) las muestras deberán ser idénticas a la presentación
más pequeña del medicamento comercializado;
e) cada muestra deberá llevar la mención "Muestra médica
gratuita - Prohibida su venta", o cualquier otra indicación de significado
análogo;
f) cada muestra deberá ir acompañada de un ejemplar del resumen
de las características del producto;
g) no podrá suministrarse muestra alguna de medicamentos que contengan
sustancias psicotrópicas o estupefacientes, con arreglo a lo definido
en los convenios internacionales, como el Convenio de las Naciones Unidas de
1961 y 1971.
2. Los Estados miembros podrán imponer mayores restricciones a la distribución
de muestras de determinados medicamentos.
Artículo 97
1. Los Estados miembros velarán por la existencia de medios adecuados
y eficaces que permitan controlar la publicidad de los medicamentos. Estos medios,
que podrán basarse en un sistema de control previo, deberán incluir
en cualquier caso disposiciones con arreglo a las cuales las personas u organizaciones
que tengan, según la legislación nacional, un interés legítimo
en la prohibición de una publicidad incompatible con el presente título
puedan interponer una acción judicial contra esta publicidad, o plantear
el caso de dicha publicidad ante un órgano administrativo competente
para decidir sobre las reclamaciones o para iniciar las correspondientes diligencias
judiciales.
2. En el marco de las disposiciones jurídicas a que se refiere el apartado
1, los Estados miembros conferirán a los tribunales o a los órganos
administrativos competencias que les faculten, en el caso de que éstos
estimen que dichas medidas son necesarias habida cuenta de todos los intereses
en juego y, en particular, del interés general:
- a ordenar el cese de una publicidad engañosa o a emprender las acciones
pertinentes con vistas a ordenar el cese de dicha publicidad,
o
- a prohibir tal publicidad o a emprender las acciones pertinentes con vistas
a ordenar la prohibición de la publicidad engañosa cuando ésta
no haya sido todavía dada a conocer al público, pero sea inminente
su publicación,
incluso en ausencia de prueba de una pérdida o de un perjuicio real,
o de una intención o negligencia por parte del anunciante.
3. Los Estados miembros dispondrán además que las medidas a que
se refiere el apartado 2 puedan ser adoptadas en el marco de un procedimiento
acelerado bien con efecto provisional, o con efecto definitivo.
Corresponderá a cada Estado miembro determinar cuál de estas dos
opciones será la que se adopte.
4. Los Estados miembros podrán otorgar a los tribunales o a los órganos
administrativos competencias que les faculten, con vistas a eliminar los efectos
persistentes de una publicidad engañosa cuyo cese haya sido ordenado
por una decisión definitiva:
- para exigir la publicación de dicha decisión total o parcialmente
y en la forma que juzguen adecuada,
- para exigir, además, la publicación de un comunicado rectificativo.
5. Los apartados 1 a 4 no excluyen el control voluntario de la publicidad de
medicamentos por parte de organismos de autorregulación y el recurso
a tales organismos, si ante los mismos pueden seguirse procedimientos, con independencia
de los procedimientos judiciales o administrativos contemplados en el apartado
1.
Artículo 98
1. El titular de la autorización de comercialización del producto
deberá crear dentro de su empresa un servicio científico encargado
de la información relativa a los medicamentos que ponga en el mercado.
2. El responsable de la comercialización:
- mantendrá a disposición de las autoridades u órganos
encargados del control de la publicidad de los productos farmacéuticos,
o bien les remitirá un ejemplar de toda publicidad emitida por su empresa,
junto con una ficha en la que se indiquen los destinatarios, el modo de difusión
y la fecha de la primera difusión;
- se asegurará de que la publicidad farmacéutica que realice su
empresa se ajuste a las prescripciones del presente título;
- verificará que los visitadores médicos empleados por su empresa
reciben la formación adecuada y respetan las obligaciones que les incumben
en virtud de los apartados 2 y 3 del artículo 93;
- proporcionará a las autoridades u órganos encargados del control
de la publicidad farmacéutica la información y la ayuda que éstos
requieran en el ejercicio de sus competencias;
- velará para que las decisiones adoptadas por las autoridades u órganos
encargados del control de la publicidad farmacéutica se respeten inmediata
e íntegramente.
Artículo 99
Los Estados miembros adoptarán las medidas necesarias para garantizar la total aplicación de todas las disposiciones del presente título y, en especial, determinarán las sanciones que se deberán imponer en caso de infracción de las disposiciones adoptadas en virtud del presente título.
Artículo 100
La publicidad de los medicamentos homeopáticos contemplados en el apartado
2 del artículo 13 y en el apartado 1 del artículo 14 se atendrá
a las disposiciones del presente título, salvo el apartado 1 del artículo
87.
No obstante, en la publicidad de dichos medicamentos sólo podrá
utilizarse la información contemplada en el apartado 1 del artículo
69.
Además, cada Estado miembro podrá prohibir en su territorio toda
publicidad de los medicamentos homeopáticos contemplados en el apartado
2 del artículo 13 y en el apartado 1 del artículo 14.
TÍTULO IX
FARMACOVIGILANCIA
Artículo 101
Los Estados miembros adoptarán todas las medidas apropiadas para fomentar
que los médicos y demás profesionales de los servicios sanitarios
informen a la autoridad competente sobre cualquier presunta reacción
adversa.
Los Estados miembros podrán obligar a los médicos y otros profesionales
sanitarios a cumplir requisitos específicos por lo que respecta a la
notificación de presuntas reacciones adversas graves o inesperadas, especialmente
cuando la notificación constituya una condición para la concesión
de una autorización de comercialización.
Artículo 102
Para asegurar la adopción de decisiones normativas adecuadas con respecto
a los medicamentos autorizados en la Comunidad, teniendo en cuenta la información
obtenida sobre reacciones adversas a los medicamentos en condiciones normales
de empleo, los Estados miembros establecerán un sistema de farmacovigilancia
para reunir información útil para la supervisión de medicamentos,
y en particular acerca de las reacciones adversas a los medicamentos en los
seres humanos, y para efectuar la evaluación científica de esa
información.
Dicha información deberá estar relacionada con los datos relativos
al consumo de los medicamentos.
Este sistema tendrá igualmente en cuenta cualquier información
relativa al uso indebido y al abuso de los medicamentos que pueda repercutir
sobre la evaluación de sus beneficios y riesgos.
Artículo 103
El titular de la autorización de comercialización tendrá
a su disposición, de manera permanente y continua, a una persona que
posea las cualificaciones apropiadas, responsable en materia de farmacovigilancia.
Esta persona cualificada estará encargada de:
a) establecer y mantener un sistema para recopilar y tratar, con el fin de que
sea accesible al menos en un lugar preciso, la información sobre todas
las presuntas reacciones adversas comunicadas al personal de la empresa y a
los visitadores médicos;
b) preparar y presentar a las autoridades competentes los informes a que se
refiere el artículo 104, en la forma exigida por dichas autoridades,
de conformidad con las orientaciones a que se refiere el apartado 1 del artículo
106;
c) asegurar que se dé una respuesta rápida y completa a toda solicitud
de información complementaria de las autoridades competentes, necesaria
para la evaluación de los beneficios y riesgos de un medicamento, incluida
la información relativa al volumen de ventas o de prescripciones del
medicamento de que se trate;
d) facilitar a las autoridades competentes cualquier otra información
de interés para la evaluación de los beneficios y riesgos asociados
a un medicamento, en particular la información relativa a estudios de
seguridad posteriores a la autorización.
Artículo 104
1. El titular de la autorización de comercialización estará
obligado a conservar informes detallados de todas las presuntas reacciones adversas
que se produzcan en el interior de la Comunidad o en un tercer país.
2. El titular de la autorización de comercialización estará
obligado a registrar y a notificar inmediatamente a la autoridad competente
del Estado miembro en cuyo territorio se haya producido el incidente, y a más
tardar dentro de los 15 días naturales siguientes a la recepción
de la información, todas las presuntas reacciones adversas graves que
le hayan sido comunicadas por profesionales sanitarios.
3. El titular de la autorización de comercialización estará
obligado a registrar y a notificar inmediatamente a la autoridad competente
del Estado miembro en cuyo territorio se haya producido el incidente, y a más
tardar dentro de los 15 días naturales siguientes a la recepción
de la información, cualquier otra presunta reacción adversa grave
que responda a los criterios de notificación con arreglo a las orientaciones
a que se refiere el apartado 1 del artículo 106 de la que pueda presumirse
razonablemente que tenga conocimiento.
4. El titular de la autorización de comercialización garantizará
que todas las presuntas reacciones adversas graves inesperadas que se produzcan
en el territorio de un tercer país, de las que tengan conocimiento a
través de un profesional de los servicios sanitarios, sean notificadas
inmediatamente de conformidad con las orientaciones a que se refiere el apartado
1 del artículo 106, de manera que estén a disposición de
la Agencia y de las autoridades competentes de los Estados miembros en los que
está autorizado el medicamento, y a más tardar dentro de los 15
días naturales siguientes a la recepción de la información.
5. En el caso de los medicamentos que entren dentro del ámbito de aplicación
de la Directiva 87/22/CEE, o que se hayan beneficiado de los procedimientos
de reconocimiento mutuo previstos en los artículos 17 y 18 de la presente
Directiva, o en el apartado 4 del artículo 28 de la presente Directiva,
o que hayan sido objeto de los procedimientos previstos en los artículos
32, 33 y 34 de la presente Directiva, el titular de la autorización de
comercialización deberá garantizar además que todas las
presuntas reacciones adversas graves producidas en la Comunidad se notifiquen
en un formato y con una periodicidad acordados con el Estado miembro de referencia,
o con una autoridad competente que actúe como Estado miembro de referencia,
de manera que sean accesibles al Estado miembro de referencia.
6. Salvo que se hayan establecido otros requisitos como condición al
conceder la autorización de comercialización, o con posterioridad,
con arreglo a las orientaciones a que se refiere el apartado 1 del artículo
106, las notificaciones de todas las reacciones adversas deberán presentarse
a la autoridad competente en forma de informe periódico de actualizado
en materia de seguridad, bien inmediatamente cuando ésta lo solicite
o bien a intervalos regulares con la siguiente periodicidad: semestralmente
durante los dos primeros años siguientes a la autorización, anualmente
durante los dos años siguientes y una vez en el momento de producirse
la primera renovación. A partir de ese momento, los informes periódicos
actualizados en materia de seguridad se presentarán a intervalos de cinco
años, junto con la solicitud de renovación de la autorización.
Los informes periódicos actualizados en materia de seguridad deberán
incluir una evaluación científica de los beneficios y riesgos
asociados al medicamento.
7. Tras la concesión de una autorización de comercialización,
su titular podrá solicitar la modificación de la periodicidad
contemplada en el presente artículo con arreglo al procedimiento establecido
por el Reglamento (CE) n° 541/95 de la Comisión(23).
Artículo 105
1. La Agencia, en colaboración con los Estados miembros y la Comisión,
establecerá una red de proceso de datos a fin de facilitar el intercambio
de información sobre farmacovigilancia relativa a los medicamentos comercializados
en la Comunidad, y que permita que todas las autoridades competentes compartan
la información al mismo tiempo.
2. Haciendo uso de la red prevista en el apartado 1, los Estados miembros velarán
por que las notificaciones sobre presuntas reacciones adversas graves que se
hayan producido en su territorio sean transmitidas a la Agencia y a los demás
Estados miembros inmediatamente, y a más tardar dentro de los 15 días
naturales siguientes a su notificación.
3. Los Estados miembros velarán por que se pongan inmediatamente a disposición
del titular de la autorización de comercialización, y a más
tardar dentro de los 15 días naturales siguientes a su notificación,
las notificaciones sobre las presuntas reacciones adversas graves que se hayan
producido en su territorio.
Artículo 106
1. A fin de facilitar el intercambio de información acerca de la farmacovigilancia
en la Comunidad, la Comisión elaborará, previa consulta a la Agencia,
a los Estados miembros y a las partes interesadas, orientaciones sobre recopilación,
verificación y presentación de informes sobre reacciones adversas,
incluyendo los requisitos técnicos en materia de intercambio electrónico
de información sobre farmacovigilancia de conformidad con las disposiciones
acordadas internacionalmente y publicarán una referencia a una terminología
médica internacionalmente aceptada.
Dichas orientaciones se publicarán en el volumen 9 de las Normas sobre
medicamentos de la Comunidad Europea y tendrán en cuenta los trabajos
de armonización internacional llevados a cabo en el ámbito de
la farmacovigilancia.
2. A efectos de la interpretación de las definiciones previstas en los
puntos 11 a 16 del artículo 1 y de los principios contenidos en el presente
título, el titular de la autorización de comercialización
y las autoridades competentes se remitirán a las orientaciones contempladas
en el apartado 1.
Artículo 107
1. Si, como resultado de la evaluación de los datos sobre farmacovigilancia,
un Estado miembro considera que una autorización previa a la comercialización
debe suspenderse, retirarse o modificarse de conformidad con las orientaciones
a que se refiere el apartado 1 del artículo 106, informará inmediatamente
de ello a la Agencia, a los demás Estados miembros y al titular de la
autorización de comercialización.
2. En casos de urgencia, el Estado miembro de que se trate podrá suspender
la autorización de comercialización de un medicamento, siempre
que informe de ello a la Agencia, a la Comisión y a los demás
Estados miembros, a más tardar el primer día hábil siguiente.
Artículo 108
Cualquier modificación que resulte necesaria para actualizar las disposiciones de los artículos 101 a 107 para tomar en consideración el progreso científico y técnico se adoptará de conformidad con el procedimiento establecido en el apartado 2 del artículo 121.
TÍTULO X
DISPOSICIONES PARTICULARES RELATIVAS A LOS MEDICAMENTOS DERIVADOS DE LA SANGRE
Y DEL PLASMA HUMANOS
Artículo 109
Para la extracción y verificación de sangre humana y de plasma humano se aplicará la Directiva 2002/98/CE del Parlamento Europeo y del Consejo, de 27 de enero de 2003, por la que se establecen normas de calidad y de seguridad para la extracción, verificación, tratamiento, almacenamiento y distribución de sangre humana y sus componentes y por la que se modifica la Directiva 2001/83/CE.
Artículo 110
Los Estados miembros adoptarán todas las medidas necesarias para promover el autoabastecimiento de la Comunidad de sangre o plasma humanos. A este respecto, deben estimularse las donaciones de sangre o de plasma voluntarias y no remuneradas y adoptarán todas las medidas necesarias para el desarrollo de la producción y de la utilización de medicamentos derivados de sangre o plasma humanos procedentes de donaciones voluntarias y no remuneradas. Comunicarán las medidas adoptadas a la Comisión.
TÍTULO XI
VIGILANCIA Y SANCIONES
Artículo 111
1. La autoridad competente del Estado miembro en cuestión se cerciorará,
mediante inspecciones reiteradas, de que se cumplen las prescripciones legales
relativas a los medicamentos.
Estas inspecciones serán efectuadas por agentes de la autoridad competente
que deberán estar facultados para:
a) proceder a inspeccionar los establecimientos fabriles y comerciales, así
como los laboratorios encargados, por el titular de la autorización de
fabricación, de efectuar controles en virtud del artículo 20;
b) tomar muestras;
c) informarse de todos los documentos relacionados con el objeto de las inspecciones,
sin perjuicio de las disposiciones en vigor en los Estados miembros a 21 de
mayo de 1975, que limitan esta facultad en lo relativo a la descripción
del modo de preparación.
2. Los Estados miembros se encargarán de que los procedimientos de fabricación
seguidos para fabricar productos farmacéuticos inmunológicos estén
validados adecuadamente y garanticen de forma continuada la conformidad de los
lotes.
3. Al término de cada una de las inspecciones mencionadas en el apartado
1, los agentes de las autoridades competentes harán un informe sobre
el cumplimiento, por parte del fabricante, de los principios y directrices de
las prácticas correctas de fabricación que establece el artículo
47. El contenido de dicho informe será comunicado al fabricante objeto
de la inspección.
Artículo 112
Los Estados miembros adoptarán todas las disposiciones adecuadas para que el titular de la autorización de comercialización y, en su caso, el titular de la autorización de fabricación, justifiquen la realización de los controles llevados a cabo sobre el medicamento y/o sobre los componentes y productos intermedios de la fabricación, según los métodos tenidos en cuenta para la autorización de comercialización mencionados en la letra h) del apartado 3 del artículo 8.
Artículo 113
A efectos de la aplicación del artículo 112, los Estados miembros podrán exigir que los fabricantes de medicamentos inmunológicos o medicamentos derivados de sangre o plasma humanos sometan a la autoridad competente copia de todos los informes de control firmados por la persona cualificada con arreglo al artículo 51.
Artículo 114
1. Cuando lo considere necesario para el interés de la salud pública,
un Estado miembro podrá exigir que el titular de la autorización
de comercialización:
- de vacunas vivas,
- de medicamentos inmunológicos utilizados para la inmunización
primaria de niños o de otros grupos de riesgo,
- de medicamentos inmunológicos utilizados en programas de inmunización
de salud pública,
- o de medicamentos inmunológicos nuevos o fabricados con técnicas
nuevas o modificadas, o que presenten un carácter de novedad para un
fabricante determinado, y ello durante un período transitorio fijado
normalmente en la autorización de comercialización,
someta al control de un laboratorio estatal o un laboratorio destinado a dicho
efecto muestras de cada lote del producto a granel y/o medicamento, antes de
su comercialización, a menos que, en el caso de un lote fabricado en
otro Estado miembro, la autoridad competente de dicho Estado miembro haya examinado
ya el lote en cuestión y lo haya declarado conforme con las especificaciones
aprobadas. Los Estados miembros velarán porque dicho examen se concluya
en los 60 días a partir de la recepción de las muestras.
2. Cuando, por razones de salud pública, la legislación de un
Estado miembro lo prevea, las autoridades competentes podrán exigir al
titular de la autorización de comercialización de medicamentos
derivados de sangre o plasma humanos la presentación ante las autoridades
competentes de muestras de cada uno de los lotes del producto a granel y/o acabado
para su examen por un laboratorio estatal o un laboratorio designado a tal fin
antes de su puesta en circulación a no ser que las autoridades competentes
de otro Estado miembro hayan examinado previamente el lote correspondiente y
hayan declarado que cumple las especificaciones aprobadas. Los Estados miembros
se encargarán de que dicho examen se realice en el plazo de sesenta días
a partir de la recepción de las muestras.
Artículo 115
Los Estados miembros se encargarán de que los procedimientos de fabricación seguidos para elaborar un medicamento derivado de sangre o plasma humanos estén validados adecuadamente, proporcionen homogeneidad entre lotes y garanticen, en la medida en que lo permita el estado de la técnica, la ausencia de contaminación vírica específica. Para ello, el fabricante deberá comunicar a las autoridades competentes el método que utiliza para reducir o eliminar los virus patógenos que puedan transmitirse por los medicamentos derivados de sangre o plasma humanos. Las autoridades competentes podrán enviar muestras del producto a granel y/o medicamento para su estudio en un laboratorio estatal o en un laboratorio designado a este efecto, bien durante el examen de la solicitud con arreglo al artículo 19 o bien después de haber concedido la autorización de comercialización.
Artículo 116
Las autoridades competentes de los Estados miembros suspenderán o retirarán
la autorización de comercialización cuando el medicamento resulte
ser nocivo en las condiciones normales de empleo, cuando carezca de efectos
terapéuticos o finalmente cuando no posea la composición cualitativa
y cuantitativa declarada. El medicamento carece de efectos terapéuticos
cuando se pruebe que mediante él no se pueden obtener resultados terapéuticos.
La autorización será igualmente suspendida o retirada cuando se
reconociera que las informaciones que figuren en el expediente en virtud del
artículo 8, del apartado 1 del artículo 10 y del artículo
11 sean erróneas o no hayan sido modificadas de conformidad con el artículo
23, o cuando los controles contemplados en el artículo 112 no hayan sido
efectuados.
Artículo 117
1. Sin perjuicio de las medidas previstas en el artículo 116, los Estados
miembros adoptarán todas las disposiciones adecuadas para que la dispensación
del medicamento sea prohibida y dicho medicamento sea retirado del mercado cuando:
a) resulte que el medicamento sea nocivo en las condiciones normales de empleo,
o
b) falte el efecto terapéutico del medicamento,
o
c) el medicamento no tenga la composición cualitativa y cuantitativa
declarada,
o
d) no hayan sido efectuados los controles sobre el medicamento producto acabado
y/o sobre los componentes y productos intermedios de la fabricación,
o cuando cualquier otra exigencia u obligación relativa al otorgamiento
de la autorización de fabricación no fuera observada.
2. La autoridad competente podrá limitar la prohibición de dispensación
y la retirada del mercado a los únicos lotes de fabricación que
fueran objeto de discusión.
Artículo 118
1. La autoridad competente suspenderá o retirará la autorización
de fabricación para una categoría de preparados o para el conjunto
de éstos cuando una de las exigencias previstas en el artículo
41 no fuera observada.
2. La autoridad competente, además de las medidas previstas en el artículo
117, podrá suspender la fabricación o importación de medicamentos
procedentes de terceros países, o bien suspender o retirar la autorización
de fabricación, para una categoría de preparados o para el conjunto
de éstos en caso de incumplimiento de los artículos 42, 46, 51
y 112.
Artículo 119
Las disposiciones del presente título se aplicarán a los medicamentos
homeopáticos, sin perjuicio de lo dispuesto en el apartado 3 del artículo
14.
TÍTULO XII
COMITÉ PERMANENTE
Artículo 120
Las modificaciones que sean necesarias para adaptar el Anexo I al progreso científico y técnico se adoptarán con arreglo al procedimiento previsto en el apartado 2 del artículo 121.
Artículo 121
1. La Comisión estará asistida por un Comité permanente
de medicamentos para uso humano para la adaptación al progreso técnico
de las directivas dirigidas a la supresión de los obstáculos técnicos
en los intercambios en el sector de los medicamentos (denominado en lo sucesivo
"Comité permanente").
2. En los casos en que se haga referencia al presente apartado, serán
de aplicación los artículos 5 y 7 de la Decisión 1999/468/CE,
observando lo dispuesto en su artículo 8.
El plazo contemplado en el apartado 6 del artículo 5 de la Decisión
1999/468/CE queda fijado en tres meses.
3. El Comité permanente aprobará su reglamento interno.
TÍTULO XIII
DISPOSICIONES GENERALES
Artículo 122
Los Estados miembros adoptarán todas las disposiciones adecuadas para
que las autoridades competentes interesadas se comuniquen mutuamente las informaciones
que procedan para garantizar la observancia de las exigencias requeridas para
la autorización de fabricación o para la autorización de
comercialización.
Tras una solicitud razonada, los Estados miembros comunicarán inmediatamente
a las autoridades competentes de otro Estado miembro los informes contemplados
en el apartado 3 del artículo 111. Si a la vista de los informes, el
Estado miembro destinatario de los informes considera que no puede aceptar las
conclusiones adoptadas por las autoridades competentes del Estado miembro en
el que se ha elaborado el informe, lo comunicará a las autoridades competentes
correspondientes exponiendo sus motivos y podrá solicitar información
suplementaria. Los Estados miembros en cuestión se esforzarán
por llegar a un acuerdo. Si fuese necesario, en caso de divergencias graves,
la Comisión será informada por uno de los Estados miembros implicados.
Artículo 123
1. Cada Estado miembro adoptará todas las disposiciones adecuadas para
que sean inmediatamente puestas en conocimiento de la Agencia las decisiones
de autorización, de comercialización, de denegación o retirada
de autorización de comercialización, de anulación de la
decisión de denegación o retirada de autorización de comercialización,
de prohibición de venta, de retirada del mercado, así como los
motivos que las justifiquen.
2. El titular de la autorización de comercialización deberá
notificar inmediatamente a los Estados miembros afectados cualquier acción
que emprendiere para suspender la comercialización o retirar el medicamento
del mercado, indicando las razones de esta acción, cuando ésta
se refiera a la eficacia del medicamento o a la protección de la salud
pública. Los Estados miembros velarán por que esta información
se ponga en conocimiento de la Agencia.
3. Los Estados miembros tomarán las medidas necesarias para que se comunique
sin demora a la Organización Mundial de la Salud una adecuada información
relativa a las acciones emprendidas de conformidad con los apartados 1 y 2 que
pudieren afectar a la protección de la salud pública en países
terceros, remitiendo una copia de ello a la Agencia.
4. La Comisión publicará cada año una lista de los medicamentos
prohibidos en la Comunidad.
Artículo 124
Los Estados miembros se comunicarán mutuamente toda la información necesaria para garantizar la calidad e inocuidad de los medicamentos homeopáticos fabricados y comercializados en la Comunidad y, en especial, la información mencionada en los artículos 122 y 123.
Artículo 125
Toda decisión de las autoridades competentes de los Estados miembros
contemplada en la presente Directiva deberá justificarse de forma precisa.
Se notificará al interesado indicándole los recursos establecidos
por la legislación vigente, así como el plazo en el que el recurso
pueda presentarse.
Los Estados miembros publicarán en sus respectivos Diarios Oficiales
las autorizaciones de comercialización y las decisiones de revocación.
Artículo 126
La autorización de comercialización sólo podrá
denegarse, suspenderse o retirarse por alguna de las causas que se enumeran
en la presente Directiva.
Toda decisión de suspensión de fabricación o de importación
de medicamentos procedentes de terceros países, así como de prohibición
de venta y de retirada de un medicamento del mercado, sólo podrá
tomarse por las razones enumeradas en los artículos 117 y 118.
Artículo 127
1. A solicitud de un fabricante, de un exportador o de las autoridades de un
país tercero importador, los Estados miembros certificarán que
un fabricante de medicamentos está en posesión de la autorización
de fabricación. Al expedir estos certificados los Estados miembros respetarán
las condiciones siguientes:
a) Tendrán en cuenta las disposiciones administrativas vigentes de la
Organización Mundial de la Salud.
b) Adjuntarán a los medicamentos destinados a la exportación,
ya autorizados en su territorio, el resumen de las características del
producto, en la forma aprobada de conformidad con el artículo 21.
2. Cuando el fabricante no esté en posesión de una autorización
de comercialización en el país de origen, presentará a
las autoridades competentes para establecer el certificado contemplado en el
apartado 1 una declaración en la que se expliquen las razones por las
que no dispone de dicha autorización.
TÍTULO XIV
DISPOSICIONES FINALES
Artículo 128
Quedan derogadas las Directivas 65/65/CEE, 75/318/CEE, 75/319/CEE, 89/342/CEE,
89/343/CEE, 89/381/CEE, 92/25/CEE, 92/26/CEE, 92/27/CEE, 92/28/CEE y 92/73/CEE,
tal y como han sido modificadas por las Directivas que figuran en la parte A
del Anexo II, sin perjuicio de las obligaciones de los Estados miembros en cuanto
a los plazos de transposición que figuran en la parte B del Anexo II.
Las referencias hechas a las directivas derogadas se entenderán hechas
a la presente Directiva, con arreglo al cuadro de correspondencias que figura
en el Anexo III.
Artículo 129
La presente Directiva entrará en vigor el vigésimo día siguiente al de su publicación en el Diario Oficial de las Comunidades Europeas.
Artículo 130
Los destinatarios de la presente Directiva son los Estados miembros.
Hecho en Bruselas, el 6 de noviembre de 2001.
Por el Parlamento Europeo
La Presidenta
N. Fontaine
Por el Consejo
El Presidente
D. Reynders
(1) DO C 368 de 20.12.1999, p. 3.
(2) Dictamen del Parlamento Europeo de 3 de julio de 2001 (no publicado aún
en el Diario Oficial) y Decisión del Consejo de 27 de septiembre de 2001.
(3) DO 22 de 9.2.1965, p. 369/65. Directiva cuya última modificación
la constituye la Directiva 93/39/CEE (DO L 214 de 24.8.1993, p. 22).
(4) DO L 147 de 9.6.1975, p. 1. Directiva cuya última modificación
la constituye la Directiva 1999/83/CE de la Comisión (DO L 243 de 15.9.1999,
p. 9).
(5) DO L 147 de 9.6.1975, p. 13. Directiva cuya última modificación
la constituye la Directiva 2000/38/CE de la Comisión (DO L 139 de 10.6.2000,
p. 28).
(6) DO L 142 de 25.5.1989, p. 14.
(7) DO L 142 de 25.5.1989, p. 16.
(8) DO L 181 de 28.6.1989, p. 44.
(9) DO L 113 de 30.4.1992, p. 1.
(10) DO L 113 de 30.4.1992, p. 5.
(11) DO L 113 de 30.4.1992, p. 8.
(12) DO L 113 de 30.4.1992, p. 13.
(13) DO L 297 de 13.10.1992, p. 8.
(14) DO L 214 de 24.8.1993, p. 1. Reglamento modificado por el Reglamento (CE)
n° 649/98 de la Comisión (DO L 88 de 24.3.1998, p. 7).
(15) DO L 265 de 5.10.1984, p. 1. Directiva derogada con efecto el 13.5.2000
por la Directiva 96/43/Euratom (DO L 180 de 9.7.1997, p. 22).
(16) DO L 246 de 17.9.1980, p. 1. Directiva modificada por la Directiva 84/467/Euratom
(DO L 265 de 5.10.1984, p. 4). Directiva derogada con efecto el 13.5.2000 por
la Directiva 96/29/Euratom (DO L 314 de 4.12.1996, p. 20).
(17) DO L 250 de 19.9.1984, p. 17. Directiva modificada por la Directiva 97/55/CE
(DO L 290 de 23.10.1997, p. 18).
(18) DO L 298 de 17.10.1989, p. 23. Directiva modificada por la Directiva 97/36/CE
(DO L 202 de 30.7.1997, p. 60).
(19) DO L 184 de 17.7.1999, p. 23.
(20) DO L 207 de 30.7.1986, p. 1.
(21) DO L 15 de 17.1.1987, p. 38. Directiva derogada por la Directiva 93/41/CEE
(DO L 214 de 24.8.1993, p. 40).
(22) DO L 187 de 9.6.1975, p. 23.
(23) DO L 55 de 11.3.1995, p. 7. Reglamento modificado por el Reglamento (CE)
n° 1146/98 (DO L 159 de 3.6.1998, p. 31).
ANEXO I
NORMAS Y PROTOCOLOS ANALÍTICOS, FARMACOTOXICOLÓGICOS Y CLÍNICOS
RELATIVOS A LA REALIZACIÓN DE PRUEBAS DE MEDICAMENTOS
Introducción y principios generales
(1) Los datos y la documentación que han de acompañar a toda
solicitud de autorización de comercialización con arreglo al artículo
8 y al apartado 1 del artículo 10 deberán presentarse según
los requisitos que se exponen en el presente anexo, siguiendo las orientaciones
publicadas por la Comisión en las Normas sobre medicamentos de la Comunidad
Europea, volumen 2 B, Nota explicativa para los solicitantes, Medicamentos de
uso humano, Presentación y contenido del expediente, Documento técnico
común (DTC).
(2) Los datos y documentos deben presentarse en cinco módulos: el módulo
1 recoge los datos administrativos específicos para la Comunidad Europea;
en el módulo 2 se incluyen los resúmenes de la calidad, clínicos
y no clínicos; el módulo 3 ofrece información química,
farmacéutica y biológica; el módulo 4 recoge los informes
no clínicos; y el módulo 5 contiene los informes de estudio clínico.
En dicha presentación se aplica un formato común para todas las
regiones de la Conferencia internacional sobre armonización (International
Conference on Harmonisation, ICH)(1): la Comunidad Europea, Estados Unidos y
Japón. Los cinco módulos mencionados han de presentarse estrictamente
con arreglo al formato, contenido y sistema de numeración que se definen
pormenorizadamente en el volumen 2 B de la mencionada Nota explicativa para
los solicitantes.
(3) La presentación del DTC de la Comunidad Europea es aplicable a todos
los tipos de solicitud de autorización de comercialización para
cualquier procedimiento que se aplique (centralizado, reconocimiento mutuo o
nacional) y tanto si se basa en una solicitud completa o simplificada. También
es aplicable a todos los tipos de productos, incluidas las Nuevas Entidades
Químicas (NEQ), radiofármacos, derivados del plasma, vacunas,
medicamentos a base de plantas, etc.
(4) Al constituir el expediente de solicitud de autorización de comercialización,
los solicitantes deberán tener asimismo en cuenta las directrices científicas
sobre calidad, seguridad y eficacia de los medicamentos de uso humano adoptadas
por el Comité de especialidades farmacéuticas y publicadas por
la Agencia Europea para la Evaluación de Medicamentos (EMEA), así
como las demás directrices farmacéuticas comunitarias publicadas
por la Comisión en los distintos volúmenes de las Normas sobre
medicamentos de la Comunidad Europea.
(5) Por lo que respecta a la parte cualitativa (química, farmacéutica
y biológica) del expediente, son aplicables la totalidad de las monografías,
incluidos los capítulos y monografías generales de la Farmacopea
europea.
(6) El proceso de fabricación deberá cumplir los requisitos de
la Directiva 91/356/CEE de la Comisión, de 13 de junio de 1991, por la
que se establecen los principios y directrices de las prácticas correctas
de fabricación de los medicamentos de uso humano(2) y con los principios
y directrices relativos a las prácticas correctas de fabricación,
publicados por la Comisión en las Normas sobre medicamentos de la Comunidad
Europea, volumen 4.
(7) Deberá incluirse en la solicitud toda la información pertinente
para la evaluación del medicamento correspondiente, tanto si resulta
favorable como desfavorable al producto. En concreto, deberán ofrecerse
todos los datos pertinentes acerca de todas las pruebas o ensayos farmacotoxicológicos
o clínicos incompletos o abandonados relativos al medicamento y/o ensayos
completos relacionados con indicaciones terapéuticas no cubiertas por
la solicitud.
(8) Todos los ensayos clínicos que se realicen en la Comunidad Europea
deberán ajustarse a los requisitos que figuran en la Directiva 2001/20/CE
del Parlamento Europeo y del Consejo, relativa a la aproximación de las
disposiciones legales, reglamentarias y administrativas de los Estados miembros
sobre la aplicación de buenas prácticas clínicas en la
realización de ensayos clínicos de medicamentos de uso humano(3).
Para poder ser tenidos en cuenta durante la evaluación de una solicitud,
los ensayos clínicos realizados fuera de la Comunidad Europea relacionados
con medicamentos destinados a ser utilizados en la misma deberán concebirse,
realizarse y notificarse, por lo que respecta a las prácticas clínicas
y principios éticos, con arreglo a principios equivalentes a los expuestos
en la Directiva 2001/20/CE. Deberán llevarse a cabo con arreglo a los
principios éticos que se recogen, por ejemplo, en la Declaración
de Helsinki.
(9) Los estudios no clínicos (farmacotoxicológicos) deberán
realizarse de acuerdo con las disposiciones sobre prácticas correctas
de laboratorio expuestas en las Directivas 87/18/CEE del Consejo, sobre la aproximación
de las disposiciones legales, reglamentarias y administrativas relativas a la
aplicación de los principios de prácticas correctas de laboratorio
y al control de su aplicación para las pruebas sobre las sustancias químicas(4)
y 88/320/CEE del Consejo, relativa a la inspección y verificación
de las buenas prácticas de laboratorio (BPL)(5).
(10) Los Estados miembros deberán garantizar también que todas
las pruebas realizadas con animales se llevan a cabo con arreglo a la Directiva
86/609/CEE del Consejo, de 24 de noviembre de 1986, relativa a la aproximación
de las disposiciones legales, reglamentarias y administrativas de los Estados
Miembros respecto a la protección de los animales utilizados para experimentación
y otros fines científicos.
(11) Con el fin de hacer un seguimiento de la evaluación de beneficios/riesgos,
deberá presentarse a la autoridad competente toda nueva información
que no figure en la solicitud original y todos los datos sobre farmacovigilancia.
Una vez concedida la autorización de comercialización, todas las
modificaciones de los datos del expediente deberán someterse a las autoridades
competentes con arreglo a los requisitos que figuran en los Reglamentos (CE)
n° 1084/2003(6) y (CE) n° 1085/2003(7) de la Comisión o, si es
pertinente, conforme a las disposiciones nacionales, así como los requisitos
expuestos en el volumen 9 de la publicación de la Comisión Normas
sobre medicamentos de la Comunidad Europea.
El presente anexo se divide en cuatro partes:
- En la parte I se expone el formato de la solicitud, el resumen de características
del producto, el etiquetado, el prospecto y los requisitos de presentación
de las solicitudes normalizadas (módulos 1 a 5).
- En la parte II se exponen las excepciones que se aplicarán a las "solicitudes
específicas", a saber: uso médico suficientemente comprobado,
medicamentos esencialmente similares, medicamentos de combinación fija,
medicamentos biológicos similares, circunstancias excepcionales y solicitudes
mixtas (parte bibliográfica y parte de estudios propios).
- En la parte III se abordan los "Requisitos particulares de las solicitudes
de autorización de comercialización" de medicamentos biológicos
(archivo principal sobre plasma; archivo principal sobre antígenos de
vacuna), radiofármacos, medicamentos homeopáticos, medicamentos
a base de plantas y medicamentos huérfanos.
- La parte IV, que trata de los "medicamentos de terapia avanzada",
aborda los requisitos específicos de los medicamentos de terapia génica
(mediante un sistema autólogo o alogénico humano, o mediante sistema
xenogénico) y medicamentos de terapia celular, tanto de origen humano
como animal, y medicamentos para trasplantes xenogénicos.
PARTE I
REQUISITOS DE LOS EXPEDIENTES NORMALIZADOS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
1.
MÓDULO 1: INFORMACIÓN ADMINISTRATIVA
1.1. Índice
Deberá presentarse un índice exhaustivo de los módulos
1 a 5 del expediente presentado para solicitar la autorización de comercialización.
1.2. Formulario de solicitud
El medicamento para el que se presenta la solicitud deberá identificarse
mediante su nombre y el nombre de la(s) sustancia(s) activa(s), junto con su
forma farmacéutica, vía de administración, dosificación
y presentación final, incluido el envase.
Deberá hacerse constar el nombre y dirección del solicitante,
así como el nombre y la dirección de los fabricantes y los lugares
donde se realizan las distintas fases de fabricación (incluido el fabricante
del producto acabado y el fabricante o fabricantes de las sustancias activas)
y, cuando proceda, el nombre y dirección del importador.
El solicitante deberá identificar el tipo de solicitud e indicar, en
su caso, las muestras que facilita.
Deberán adjuntarse con los datos administrativos copias de la autorización
de fabricación que se define en el artículo 40, junto con una
lista de países en los que se ha concedido la autorización, copias
de los resúmenes de características del producto aprobadas por
los Estados miembros y la lista de países en los que se ha presentado
la solicitud.
Tal como se señala en el formulario de solicitud, los solicitantes deberán
facilitar, entre otros elementos, datos detallados sobre el medicamento objeto
de la misma, el fundamento jurídico de la solicitud, el titular propuesto
de la autorización de comercialización y el fabricante o fabricantes,
información sobre la situación jurídica de los medicamentos
huérfanos, dictámenes científicos y un programa de desarrollo
pediátrico.
1.3. Resumen de las características del producto, etiquetado y prospecto
1.3.1. Resumen de las características del producto
El solicitante deberá proponer un resumen de las características
del producto, con arreglo al artículo 11.
1.3.2. Etiquetado y prospecto
Deberá facilitarse el texto de etiquetado propuesto para el acondicionamiento
primario y el embalaje exterior, así como para el prospecto. Todos ellos
deberán ajustarse a los elementos obligatorios que se relacionan en el
título V sobre el etiquetado de los medicamentos de uso humano (artículo
63) y sobre el prospecto (artículo 59).
1.3.3. Maquetas y muestras
El solicitante deberá facilitar muestras y/o maquetas del acondicionamiento
primario y del embalaje exterior, las etiquetas y los prospectos del medicamento
correspondiente.
1.3.4. Resúmenes de las características del producto ya aprobados
en el Estado miembro
Con los datos administrativos del formulario de solicitud se adjuntarán
copias de todos los resúmenes de características del producto
con arreglo a los artículos 11 y 21 tal como los hayan aprobado los Estados
miembros, en su caso, y una lista de países en los que se ha presentado
solicitud.
1.4. Información acerca de los expertos
Con arreglo al apartado 2 del artículo 12, los expertos deberán
facilitar informes detallados de sus comprobaciones sobre los documentos y los
datos que constituyen el expediente de autorización de comercialización,
en concreto los módulos 3, 4 y 5 (documentación química,
farmacéutica y biológica, documentación no clínica
y documentación clínica, respectivamente). Los expertos deberán
abordar los puntos decisivos relacionados con la calidad del medicamento y de
los estudios realizados en animales y seres humanos y notificar todos los datos
pertinentes para la evaluación.
Estos requisitos deberán cumplirse facilitando un resumen global de la
calidad, una visión general de la parte no clínica (datos extraídos
de estudios realizados en animales) y una visión general de la parte
clínica que se incluirá en el módulo 2 del expediente de
solicitud de autorización de comercialización. En el módulo
1 se presentará una declaración firmada por los expertos, junto
con una síntesis de sus datos académicos, su formación
y su experiencia laboral. Los expertos deberán poseer la adecuada cualificación
técnica o profesional. Deberá declararse la relación profesional
entre el experto y el solicitante.
1.5. Requisitos especiales para los distintos tipos de solicitudes
En la parte II del presente anexo se exponen los requisitos específicos
para los distintos tipos de solicitudes.
1.6. Evaluación del riesgo para el medio ambiente
Si procede, en las solicitudes de autorización de comercialización
se incluirá una evaluación general de los posibles riesgos para
el medio ambiente debido a la utilización y/o eliminación del
medicamento y se formularán las propuestas de disposiciones relativas
al etiquetado que procedan. Deberán abordarse los riesgos para el medio
ambiente relacionados con la liberación de medicamentos que contengan
o consistan en organismos modificados genéticamente (OMG) con arreglo
al artículo 2 de la Directiva 2001/18/CE del Parlamento Europeo y del
Consejo de 12 de marzo de 2001 sobre la liberación intencional en el
medio ambiente de organismos modificados genéticamente y por la que se
deroga la Directiva 90/220/CEE del Consejo(8).
La información relacionada con el riesgo para el medio ambiente deberá
figurar como anexo del módulo 1.
La información se presentará con arreglo a lo dispuesto en la
Directiva 2001/18/CE, teniendo en cuenta todos los documentos de orientación
publicados por la Comisión acerca de la aplicación de la mencionada
Directiva.
La información constará de los elementos siguientes:
- una introducción;
- una copia de todos los posibles consentimientos por escrito a la liberación
intencional en el medio ambiente de OMG con fines de investigación y
desarrollo con arreglo a la parte B de la Directiva 2001/18/CE;
- la información que se exige en los anexos II a IV de la Directiva 2001/18/CE,
incluidos los métodos de detección e identificación y el
identificador único de los OMG, más toda información suplementaria
sobre los OMG o el producto que resulte pertinente para la evaluación
del riesgo para el medio ambiente;
- un informe de evaluación del riesgo para el medio ambiente elaborado
a partir de la información que se especifica en los anexos III y IV de
la Directiva 2001/18/CE y con arreglo al anexo II de la Directiva 2001/18/CE;
- una conclusión en la que se tenga en cuenta la información anterior
y la evaluación del riesgo para el medio ambiente y se proponga una estrategia
adecuada de gestión de riesgos que incluya, en lo que concierne a los
OMG y el producto correspondiente, un plan de seguimiento de la fase de poscomercialización
y la determinación de toda indicación especial que deba figurar
en el resumen de características del producto, el etiquetado o el prospecto;
- medidas adecuadas para informar a los ciudadanos.
Deberá incluirse la firma con fecha del autor, los datos académicos,
de formación y experiencia laboral del autor y una declaración
de la relación entre el autor y el solicitante.
2.
MÓDULO 2: RESÚMENES
El objeto del presente módulo es resumir los datos químicos,
farmacéuticos y biológicos y los datos no clínicos y clínicos
presentados en los módulos 3, 4 y 5 del expediente de autorización
de comercialización, y proporcionar los informes y síntesis descritos
en el artículo 12 de la presente Directiva.
Deberán tratarse y analizarse los puntos decisivos. Se ofrecerán
resúmenes objetivos en los que se incluirán tablas. En los informes
se remitirá a las tablas o a la información que contenga la documentación
principal presentada en el módulo 3 (documentación química,
farmacéutica y biológica), el módulo 4 (documentación
no clínica) y el módulo 5 (documentación clínica).
La información que contenga el módulo 2 deberá presentarse
con arreglo al formato, contenido y sistema de numeración que se definen
en el volumen 2 de la Nota explicativa para los solicitantes. Las síntesis
y resúmenes deberán ajustarse a los principios y requisitos básicos
que se establecen a continuación:
2.1. Índice general
En el módulo 2 deberá figurar un índice de la documentación
científica presentada en los módulos 2 a 5.
2.2. Introducción
Deberá indicarse la clase farmacológica, el modo de acción
y la utilización clínica propuesta del medicamento para el que
se solicita la autorización de comercialización.
2.3. Resumen global de la calidad
Se presentará un resumen global de la calidad, en el que se examinará
la información relacionada con los datos químicos, farmacéuticos
y biológicos.
Deberá hacerse hincapié en los parámetros críticos
y cuestiones fundamentales en relación con aspectos de calidad, así
como en la justificación de los casos en los no que se sigan las directrices
pertinentes. En este documento se expondrán las líneas generales
de los datos detallados correspondientes que se presentan en el módulo
3.
2.4. Visión general de la parte no clínica
Deberá presentarse una valoración integrada y crítica
de la evaluación no clínica del medicamento en animales/in vitro.
Deberá incluirse la discusión y justificación de la estrategia
de ensayo y de la desviación respecto a las directrices pertinentes.
Excepto para los medicamentos biológicos, deberá incluirse una
evaluación de las impurezas y productos de degradación, así
como de sus potenciales efectos farmacológicos y toxicológicos.
Deberán discutirse las repercusiones de cualquier posible diferencia
en la quiralidad, la forma química y el perfil de impurezas entre el
compuesto utilizado en los estudios no clínicos y el producto que se
desea comercializar.
Para los medicamentos biológicos, se evaluará la comparabilidad
del material utilizado en los estudios no clínicos, los estudios clínicos
y el medicamento que se desea comercializar.
Deberá realizarse una evaluación específica de la seguridad
de todo nuevo excipiente.
Se definirán las características del medicamento demostradas en
los estudios no clínicos y se discutir las repercusiones de las conclusiones
en relación con la seguridad del medicamento para la utilización
clínica prevista en el ser humano.
2.5. Visión general de la parte clínica
La visión general de la parte clínica tiene por objeto ofrecer
un análisis crítico de los datos clínicos incluidos en
el resumen clínico y el módulo 5. Se expondrán el enfoque
del desarrollo clínico del medicamento, incluyendo el diseño del
estudio crítico, las decisiones relacionadas con los estudios y la realización
de los mismos.
Se ofrecerá una breve visión general de las conclusiones clínicas,
en la que se tratarán las limitaciones importantes y se evaluarán
los riesgos y beneficios a partir de las conclusiones de los estudios clínicos.
Deberá interpretarse de qué modo las conclusiones relativas a
la eficacia y a la seguridad justifican la dosis propuesta y las indicaciones
y una evaluación de cómo el resumen de características
del producto y otros optimizarán los beneficios y afrontarán los
riesgos.
Se expondrán las cuestiones relativas a la eficacia o la seguridad que
se planteen en el desarrollo, así como los problemas pendientes de resolución.
2.6. Resumen no clínico
Los resultados de los estudios de farmacología, farmacocinética
y toxicología realizados en animales/in vitro se presentarán como
resúmenes objetivos escritos y tabulados, que se presentarán en
el orden siguiente:
- Introducción
- Resumen escrito de farmacología
- Resumen tabulado de farmacología
- Resumen escrito de farmacocinética
- Resumen tabulado de farmacocinética
- Resumen escrito de toxicología
- Resumen tabulado de toxicología.
2.7. Resumen clínico
Se ofrecerá un resumen objetivo detallado de la información clínica
relativa al medicamento que se incluye en el módulo 5. Comprenderá
los resultados de todos los estudios biofarmacéuticos, de los estudios
clínicos de farmacología y de los estudios clínicos sobre
eficacia y seguridad. Deberá presentarse una sinopsis de cada estudio.
La información clínica resumida se presentará en el orden
siguiente:
- Resumen de los estudios biofarmacéuticos y los métodos analíticos
relacionados
- Resumen de los estudios clínicos de farmacología
- Resumen sobre eficacia clínica
- Resumen sobre seguridad clínica
- Sinopsis de cada estudio.
3. MÓDULO
3: INFORMACIÓN QUÍMICA, FARMACÉUTICA Y BIOLÓGICA
PARA MEDICAMENTOS QUE CONTENGAN SUSTANCIAS ACTIVAS QUÍMICAS Y/O BIOLÓGICAS
3.1. Formato y presentación
El esquema general del módulo 3 es el siguiente:
- Índice
- Conjunto de datos
- Prtincipio activo
Información general
- Nomenclatura
- Estructura
- Propiedades generales
Fabricación
- Fabricante(s)
- Descripción del proceso de fabricación y de los controles en
proceso
- Control de materiales
- Control de las etapas críticas y los productos intermedios
- Validación y/o evaluación del proceso
- Desarrollo del proceso de fabricación
Caracterización
- Elucidación de la estructura y otras características
- Impurezas
Control del principio activo
- Especificaciones
- Procedimientos analíticos
- Validación de los procedimientos analíticos
- Análisis de lotes
- Justificación de las especificaciones
Estándares o materiales de referencia
Sistema de cierre del envase
Estabilidad
- Resumen y conclusiones sobre estabilidad
- Protocolo de estabilidad después de la autorización y compromiso
de estabilidad
- Datos de estabilidad
- Producto terminado
Descripción y composición del medicamento
Desarrollo farmacéutico
- Componentes del medicamento
- Principio activo
- Excipientes
- Medicamento
- Desarrollo de la formulación
- Sobredosificación
- Propiedades físico-químicas y biológicas
- Desarrollo del proceso de fabricación
- Sistema de cierre del envase
- Atributos microbiológicos
- Compatibilidad
Fabricación
- Fabricante(s)
- Fórmula del lote
- Descripción del proceso de fabricación y de los sistemas de
control del proceso
- Control de etápas críticas y de los productos intermedios
- Validación y/o evaluación del proceso
Control de los excipientes
- Especificaciones
- Procedimientos analíticos
- Validación de los procedimientos analíticos
- Justificación de las especificaciones
- Excipientes de origen humano o animal
- Nuevos excipientes
Control del producto terminado
- Especificación(-ones)
- Procedimientos analíticos
- Validación de los procedimientos analíticos
- Análisis de lotes
- Caracterización de las impurezas
- Justificación de la especificación(-ones)
Estándares o materiales de referencia
Sistema de cierre del envase
Estabilidad
- Resumen y conclusiones sobre estabilidad
- Protocolo de estabilidad después de la autorización y compromiso
de estabilidad
- Datos de estabilidad
- Anexos
- Instalaciones y equipo (únicamente medicamentos biológicos)
- Evaluación de la seguridad respecto a los agentes extraños/externos
- Excipientes
- Información suplementaria para la Comunidad Europea
- Esquema de la validación del proceso para el producto terminado
- Producto sanitario
- Certificado(s) de idoneidad
- Medicamentos que contengan o utilicen en el proceso de fabricación
materiales de origen animal y/o humano (procedimiento relativo a las encefalopatías
espongiformes transmisibles, EET)
- Referencias bibliográficas.
3.2. Contenido: principios básicos y requerimientos
(1) En los datos químicos, farmacéuticos y biológicos
que se faciliten deberán incluir, en relación con el (los) pricipio(s)
activo(s) y el producto terminado, toda la información pertinente acerca
del desarrollo, el proceso de fabricación, la caracterización
y propiedades, operaciones y requisitos de control de calidad estabilidad, así
como una descripción de la composición y presentación del
producto terminado.
(2) Se presentarán dos conjuntos principales de datos, respectivamente
relacionados con, el (los) principio(s) activo(s) y con el producto terminado.
(3) Este módulo deberá, además proporcionar información
detallada sobre los materiales de partida y materias primas utilizados durante
las operaciones de fabricación del principio(s) activo(s), y los excipientes
incorporados en la formulación del producto terminado.
(4) Todos los procedimientos y métodos utilizados para la fabricación
y control del principio activo y el producto terminado deberán describirse
de manera suficientemente pormenorizada para que puedan reproducirse en los
ensayos realizados a petición de la autoridad competente. Todos los ensayos
estarán en consonancia con el estado actual del progreso científico
y deberán estar validados. Se proporcionarán los resultados de
los estudios de validación. En el caso de los procedimientos de ensayo
incluidos en la Farmacopea Europea, esta descripción deberá sustituirse
por la referencia correspondiente a la(s) monografía(s) y capítulo(s)
general(es).
(5) Las monografías de la Farmacopea Europea deberán ser aplicables
a todas las sustancias, preparados y formas farmacéuticas que figuren
en ella Con respecto a otras substancias, cada Estado Miembro podría
solicitar el cumplimiento con su farmacopea nacional.
No obstante, cuando un material de la Farmacopea Europea o de la farmacopea
de un Estado Miembro haya sido preparado mediante un método susceptible
de dejar impurezas no controladas en la monografía de la farmacopea,
se deberán declarar dichas impurezas y sus límites máximos
de tolerancia y deberá describirse un procedimiento de ensayo adecuado.
En aquellos casos en que una especificación que figure en una monografía
de la Farmacopea Europea o en la farmacopea de un Estado Miembro pueda resultar
insuficiente para garantizar la calidad de la sustancia, las autoridades competentes
podrán solicitar al titular de la autorización de comercialización
especificaciones más adecuadas. Las autoridades competentes deberán
informar a las autoridades responsables de la farmacopea de que se trate. El
titular de la autorización de comercialización proporcionará
a las autoridades responsables de dicha farmacopea los detalles de la presunta
insuficiencia y las especificaciones adicionales aplicadas.
En el caso de los procedimientos analíticos incluidos en la Farmacopea
europea, podrá sustituirse tal descripción en cada apartado pertinente
por la referencia pormenorizada que proceda a la(s) monografía(s) y capítulo(s)
general(es).
(6) En caso de que los materiales de partida, materias primas, principi(s)
activo(s) o excipiente(s) no estén descritos en la Farmacopea Europea
ni en la farmacopea de un Estado Miembro, podrá aceptarse el cumplimiento
con la monografía de la farmacopea de un tercer país. En estos
casos, el solicitante presentará una copia de la monografía, acompañada
por la validación de los procedimientos analíticos contenidos
en la monografía y, por una traducción, cuando proceda.
(7) En caso de que el principio activo y/o material de partida, materia prima
o los excipientes sean objeto de una monografía de la Farmacopea Europea,
el solicitante podrá hacer referencia a un certificado de idoneidad,
que, cuando haya sido expedido por la Dirección Europea para la Calidad
del Medicamento (EDQM), se presentará en el apartado que corresponda
del presente Módulo. Se considerará que dichos certificados de
idoneidad de la monografía de la Farmacopea europea sustituyen los datos
pertinentes de los apartados correspondientes descritos en este módulo.
El fabricante garantizará por escrito al solicitante que el proceso de
fabricación no se ha modificado desde la concesión del certificado
de idoneidad por parte de la Dirección Europea para la Calidad del Medicamento.
(8) Para un principio activo bien definido, su fabricante o el solicitante
podrán disponer que:
(i) la descripción del proceso de fabricación,
(ii) el control de calidad durante la fabricación, y
(iii) la validación del proceso
se faciliten en un documento separado (parte cerrada) dirigido directamente
a las autoridades competentes por el fabricante del principio activo, en calidad
de archivo maestro del principio activo.
En este caso, sin embargo, el fabricante deberá proporcionar al solicitante
todos los datos que puedan resultar necesarios para que este asuma la responsabilidad
del medicamento. El fabricante deberá comprometerse por escrito frente
al solicitante a garantizar la homogeneidad de los lotes y a no modificar el
proceso de fabricación o las especificaciones sin haberle previamente
informado. Se deberán presentar a las autoridades competentes los documentos
en apoyo de esta solicitud de modificación; dichos documentos también
se proporcionarán al solicitante cuando se refieran a la parte abierta
del archivo maestro.
(9) Medidas específicas concernientes a la prevención de la transmisión
de encefalopatías espongiformes animales (materiales procedentes de rumiantes):
en cada fase del proceso de fabricación, el solicitante deberá
demostrar el cumplimiento de los materiales utilizados con la Nota Explicativa
sobre Minimización del Riesgo de Transmisión de los Agentes de
la Encefalopatía Espongiforme Animal a través de medicamentos
y sus actualizaciones, publicada por la Comisión en el Diario Oficial
de la Unión Europea. La demostración del cumplimiento con lo dispuesto
en la mencionada Nota Explicativa, podrá realizarse presentando preferiblemente,
un certificado de idoneidad en relación con la monografía pertinente
de la Farmacopea europea expedido por la Dirección Europea para la Calidad
del Medicamento, o bien los datos científicos que corroboren dicho cumplimiento.
(10) En relación con los agentes extraños/externos, deberá
facilitarse información que evalúe el riesgo con respecto a la
contaminación potencial por dicho tipo de agentes, bien sean virales
o no virales, tal como se establece en las directrices correspondientes y en
la monografía y el capítulo generales pertinentes de la Farmacopea
Europea.
(11) Se describirán con los detalles necesarios todos los aparatos y
equipos especiales que puedan utilizarse en alguna fase del proceso de fabricación
y las operaciones de control del producto terminado.
(12) En los casos en que proceda y sea necesario, se presentará la marca
CE requerida por la legislación comunitaria sobre productos sanitarios.
Deberá prestarse particular atención a los elementos seleccionados
siguientes.
3.2.1. Principio o principios activos
3.2.1.1. Información general e información sobre los materiales
de partida y materias primas
a) Se proporcionará información sobre la nomenclatura del principio
activo incluyendo la denominación común internacional recomendada
(DCI), la denominación de la Farmacopea Europea si procede y la(s) denominación(-ones)
química(s).
Se proporcionará la fórmula estructural, incluyendo la estereoquímica
relativa y absoluta, la fórmula molecular y la masa molecular relativa.
En el caso de los medicamentos biotecnológicos, si procede, deberá
comunicarse la secuencia de aminoácidos esquemática y la masa
molecular relativa.
Se presentará una lista de propiedades físico-químicas
y otras propiedades relevantes de la sustancia activa, incluyendo la actividad
biológica en el caso de los medicamentos biológicos.
b) A efectos del presente anexo, se entenderá por materiales de partida
todos los materiales a partir de los cuales se fabrica o de los que se extrae
el principio activo.
En el caso de los medicamentos biológicos, se entenderá por materiales
de partida toda sustancia de origen biológico, tales como los microorganismos,
órganos y tejidos de origen vegetal o animal, las células o fluidos
(incluyendo sangre y plasma) de origen humano o animal y los diseños
celulares biotecnológicos (sustratos celulares, sean o no recombinantes,
incluidas las células primarias).
Un medicamento biológico es un producto cuyo principio activo es biológico.
Una sustancia biológica es aquélla que se produce o se extrae
a partir de una fuente biológica y que necesita, para su caracterización
y determinación de su calidad, una combinación de ensayos fisico-químico
y biológico junto con el proceso de producción y su control. Se
considerarán medicamentos biológicos: los medicamentos inmunológicos
y los medicamentos derivados de la sangre o el plasma humanos, tal y como se
definen en los apartados 4 y 10 del artículo 1; los medicamentos que
pertenezcan al ámbito de aplicación de la parte A del anexo del
Reglamento (CEE) n° 2309/93; los medicamentos de terapia avanzada, definidos
en la parte IV de este anexo.
Cualquier otra substancia utilizada para la fabricación o extracción
del (los) principio(s) activo(s), pero de las que no deriva directamente dicho
principio activo, como los reactivos, los medios de cultivo, suero de ternera
fetal, aditivos y soluciones tampón utilizadas para la cromatografía,
etc., se consideran materias primas.
3.2.1.2. Proceso de fabricación del principio o principios activos
a) La descripción del proceso de fabricación del principio activo
representa el compromiso del solicitante respecto a la fabricación del
principio activo. Para describir de manera adecuada el proceso de fabricación
y los controles en proceso, se facilitará la información adecuada
que se establece en las directrices publicadas por la Agencia.
b) Se presentará una relación de todos los materiales necesarios
para fabricar el (los) principio(s) activo(s), identificando en qué parte
del proceso se utiliza cada material. Se facilitará información
sobre la calidad y el control de dichos materiales. También se presentará
información que demuestre que los materiales cumplen los estándares
apropiados para su utilización prevista.
Se presentará una relación de las materias primas y se documentarán
también su calidad y sus procedimientos de control.
Se proporcionarán el nombre, la dirección y la responsabilidad
de cada fabricante, incluyendo sus contratistas, y cada una de las sedes de
producción o instalaciones propuestas dedicadas a la fabricación
y control.
c) Para los medicamentos biológicos se aplicarán los siguientes
requisitos adicionales.
Se describirá y documentará el origen y la historia de los materiales
de partida.
Respecto a las medidas específicas de prevención de la transmisión
de las encefalopatías espongiformes animales, el solicitante deberá
demostrar que el principio activo cumple con lo dispuesto en la Nota Explicativa
sobre Minimización del Riesgo de Transmisión de los Agentes de
la Encefalopatía Espongiforme Animal a través de Medicamentos
y sus actualizaciones, publicada por la Comisión en el Diario Oficial
de la Unión Europea.
Cuando se usen bancos celulares, deberá demostrarse que las características
de las células se han mantenido inalteradas en los pasos empleados para
la producción y posteriormente.
Los materiales de siembra, los bancos de células, las mezclas de suero
o plasma sin elaborar y demás materias de origen biológico, así
como, siempre que sea posible, los materiales de los que se hayan obtenido,
deberán someterse a ensayos para comprobar que están libres de
agentes extraños/externos.
Cuando la presencia de agentes extraños/externos potencialmente patogénicos
es inevitable, el material correspondiente deberá utilizarse si un tratamiento
posterior garantiza su eliminación y/o inactivación, y esto deberá
ser validado.
Siempre que sea posible, la producción de vacunas deberá basarse
en un sistema de lotes de siembra y de bancos celulares establecidos. En el
caso de vacunas bacterianas y virales, las características del agente
infeccioso deberán demostrarse en los materiales de siembra. Además,
para las vacunas vivas, la estabilidad de las características de atenuación
deberán ser demostradas en el material de siembra, si esta prueba no
es suficiente, las características de atenuación deberán
también demostrarse en la etapa de producción.
Cuando se trate de medicamentos derivados de la sangre o del plasma humano,
deberán describirse y documentarse, con arreglo a lo dispuesto en la
parte III del presente anexo el origen y los criterios de recogida, transporte
y conservación de los materiales de partida.
Se describirán las instalaciones y el equipo de fabricación.
d) Deberán facilitarse, si procede, los ensayos y los criterios de aceptación
realizados en cada una de las etapas críticas, información sobre
la calidad y el control de los productos intermedios, así como sobre
la validación del proceso y/o los estudios de evaluación.
e) Si la presencia de agentes extraños/externos potencialmente patógenos,
es inevitable, el material correspondiente deberá utilizarse únicamente
si un tratamiento posterior garantiza su eliminación y/o inactivación,
y esto deberá ser validado en el apartado en que se aborde la evaluación
de la seguridad viral.
f) Se facilitará una descripción y discusión de los cambios
significativos introducidos en el proceso de fabricación durante el desarrollo
y/o el lugar de fabricación del principio activo.
3.2.1.3. Caracterización del principio o principios activos
Deberán presentarse datos que pongan de manifiesto la estructura y otras
características de la(s) sustancia(s) activa(s).
Se facilitará la confirmación de la estructura de la(s) sustancia(s)
activa(s) a partir de algún método físico-químico
y/o inmuno-químico y/o biológico, así como información
sobre las impurezas.
3.2.1.4. Control del principio(s) activo(s)
Se presentará información detallada sobre las especificaciones
utilizadas para los controles de rutina del (los) principio(s) activo(s), la
justificación de la elección de dichas especificaciones, métodos
de análisis y su validación.
Se presentarán los resultados del control efectuado en lotes fabricados
durante el desarrollo.
3.2.1.5. Estándares o materiales de referencia
Se identificarán y describirán detalladamente los estándares
y preparaciones de referencia. Cuando sea relevante, se utilizará material
de referencia químico y biológico de la Farmacopea Europea.
3.2.1.6. Envase y sistema de cierre de la sustancia activa
Se presentará la descripción del envase y el sistema o sistemas
de cierre y sus especificaciones.
3.2.1.7. Estabilidad del principio(s) activo(s)
a) Deberán resumirse los tipos de estudios realizados, los protocolos
empleados y los resultados de los estudios.
b) Se presentarán con el formato adecuado los resultados detallados de
los estudios de estabilidad, incluyendo información relativa a los procedimientos
de analíticos para obtener dichos datos, así como la validación
de estos procedimientos.
c) Se facilitarán el protocolo de estabilidad tras la autorización
y el compromiso de estabilidad.
3.2.2. Producto terminado
3.2.2.1. Descripción y composición del producto terminado
Deberá describirse el producto terminado y su composición. La
información deberá incluir la descripción de la forma farmacéutica
y su composición con todos los componentes del producto terminado, la
cantidad de los mismos por unidad y la función:
- del principio(s) activo(s),
- componente(s) los excipientes, cualquiera que sea su naturaleza o la cantidad
utilizada, incluyendo los colorantes, conservantes, adyuvantes, estabilizadores,
espesantes, emulsionantes, correctores del sabor, aromatizantes, etc.,
- los componentes de la cubierta externa de los medicamentos (cápsulas
duras, cápsulas blandas, supositorios, comprimidos recubiertos, comprimidos
recubiertos con cubierta películar, etc.) que vayan a ser ingeridos o
administrados al paciente de otra forma.
- Estos aspectos deberán completarse con cualquier otro dato revelante
relacionado con el tipo de envase y, si procede, su sistema de cierre, junto
con detalle de los dispositivos que serán utilizados para la administración
del medicamento y que se suministrarán con él.
De acuerdo con lo dispuesto en la letra c) del apartado 3 del artículo
8, la "terminología usual", a utilizar en la descripción
de los componentes del medicamento, deberá ser:
- cuando se trate de productos que figuren en la Farmacopea europea o, en su
defecto, en la farmacopea nacional de un Estado Miembro, la denominación
principal recogida en el encabezamiento de la correspondiente monografía
con referencia a la farmacopea de que se trate;
- para los restantes productos, la denominación común internacional
recomendada por la Organización Mundial de la Salud o, en su defecto,
la denominación científica exacta; las sustancias que carezcan
de denominación común internacional o de denominación científica
exacta se describirán declarando a su origen y modo de obtención,
completándose estos datos con cualquier otro detalle relevante, en caso
necesario;
- con respecto a los colorantes, la designación por el código
"E" que se les atribuya en la Directiva 78/25/CEE del Consejo, de
12 de diciembre de 1977, relativa a la aproximación de las legislaciones
de los Estados Miembros referentes a los colorantes autorizados para el uso
en los medicamentos(9) y/o en la Directiva 94/36/CE del Parlamento Europeo y
del Consejo, de 30 de junio de 1994, relativa a los colorantes utilizados en
los productos alimenticios(10).
Para proporcionar la "composición cuantitativa" de los principioas
activos del medicamento, será preciso, según la forma farmacéutica,
especificar la masa o el número de unidades de actividad biológica,
bien sea por dosis o por unidad de masa o de volumen, de cada sustancia activa.
Los principios activos presentes en forma de compuestos o derivados se designarán
cuantitativamente mediante su masa total y, en caso necesario o relevante, mediante
la masa de las fracciones activas de la molécula.
En el caso de los medicamentos que contengan un principio activo cuya autorización
se haya solicitado en cualquier Estado miembro por primera vez, la declaración
cuantitativa de un principio activo que sea una sal o hidrato se expresará
sistemáticamente en términos de masa de los fragmentos activos
de la molécula. Todas las autorizaciones posteriores de medicamentos
en los Estados miembros dispondrán de su composición cualitativa
expresada de la misma manera para el mismo principio activo.
Las unidades de actividad biológica se emplearán en las sustancias
que no pueden definirse en términos moleculares. Cuando la Organización
Mundial de la Salud haya definido una unidad de actividad biológica,
es ésta la que deberá usarse. En los casos en los que no se haya
definido una unidad internacional, las unidades de actividad biológica
se expresarán de forma que proporcionen información inequívoca
sobre la actividad de la sustancia, utilizando, cuando proceda, las unidades
de la Farmacopea europea.
3.2.2.2. Desarrollo farmacéutico
El presente capítulo se dedicará a la información sobre
los estudios de desarrollo realizados para establecer que la forma farmacéutica,
la formulación, el proceso de fabricación, el sistema de cierre
del envase, los atributos microbiológicos y las instrucciones de uso
son los adecuados para el uso previsto especificado en el expediente de solicitud
de autorización de comercialización.
Los estudios descritos en el presente capítulo son distintos de las pruebas
de controles de rutina que se realizan según las especificaciones. Se
determinarán y describirán los parámetros críticos
de la formulación y los atributos del proceso que puedan influir en la
reproducibilidad del lote, la eficacia del medicamento y su calidad. Los datos
de apoyo adicionales, deberán remitir, cuando proceda a los capítulos
relevantes del Módulo 4 (Informes de estudios no clínicos) y del
Módulo 5 (Informes de estudios clínicos) del expediente de solicitud
de autorización de comercialización.
a) Deberá documentarse la compatibilidad del principio activo con los
excipientes, así como las características físico-químicas
clave del principio activo que puedan influir en la eficacia del producto terminado
o en la compatibilidad de los distintos principios activos entre sí en
el caso de los productos en los que se combinen.
b) Se documentará la elección de los excipientes, especialmente
en relación con sus funciones respectivas y su concentración.
c) Se describirá el desarrollo del producto terminado, teniendo en consideración
la vía de administración y la utilización propuestas.
d) Deberá justificarse cualquier sobredosificación en la formulación(ones).
e) En lo que respecta a las propiedades físico-químicas y biológicas,
deberán tratarse y documentarse todos los parámetros que conciernan
al comportamiento del producto terminado.
f) Se presentará la selección y optimización del proceso
de fabricación, así como las diferencias entre el (los) proceso(s)
de fabricación utilizados para producir lotes clínicos pivotales
y el proceso empleado para la fabricación del producto terminado propuesto.
g) Se documentará la idoneidad del envase y el sistema de cierre empleado
para el almacenamiento, el transporte y la utilización del producto terminado.
Puede ser necesario considerar la posible interacción entre el medicamento
y el envase.
h) Los atributos microbiológicos de la forma farmacéutica en relación
con productos no estériles y estériles deberán ajustarse
a lo prescrito en la Farmacopea Europea y documentarse con arreglo a ello.
i) Con el fin de ofrecer información útil y adecuada para el etiquetado,
se documentará la compatibilidad del producto terminado con los diluyentes
de reconstitución y los dispositivos de administración.
3.2.2.3. Proceso de fabricación del producto terminado
a) La descripción del método de fabricación que, conforme
a lo establecido en la letra d) del apartado 3 del artículo 8, deberá
acompañar a la solicitud de autorización se redactará de
forma que ofrezca una idea clara del carácter de las operaciones efectuadas.
Con este fin, dicha descripción deberá incluir, como mínimo:
- referencia a las diferentes fases del proceso de fabricación, incluidos
los sistemas de control del proceso y los criterios de aceptación correspondientes,
de modo que se pueda evaluar si los procesos utilizados en la producción
de la forma farmacéutica puedan producir un cambio adverso en los componentes;
- en caso de fabricación en serie, información completa sobre
las precauciones tomadas para asegurar la homogeneidad del producto terminado;
- estudios experimentales de validación del procedimiento de fabricación
cuando se emplee un método de fabricación no estándar o
cuando sea crítico para el producto;
- en el caso de medicamentos estériles, detalles de los procesos de esterilización
y/o asépticos utilizados;
- la fórmula detallada del lote.
Se presentará el nombre, la dirección y la responsabilidad de
cada fabricante, incluidos sus contratistas, y cada una de las sedes de producción
o instalaciones propuestas dedicadas a la fabricación y ensayo.
b) Se incluirán los datos relativos a los ensayos de control del producto
que puedan realizarse en una fase intermedia del proceso de fabricación,
con el fin de asegurar la consistencia de la producción.
Estos ensayos son esenciales para comprobar la conformidad del medicamento con
la fórmula cuando, excepcionalmente el solicitante proponga, un método
analítico para analizar el producto terminado que no incluya la determinación
de todas los principios activos (o de todos los componentes del excipiente sujetos
a los mismos requerimientos que las sustancias activas).
Lo anterior será igualmente aplicable cuando el control de calidad del
producto terminado dependa de los controles en proceso, especialmente en el
caso de que el medicamento se defina principalmente por su proceso de preparación.
c) Se presentará descripción, documentación y resultados
de los estudios de validación para las etapas o ensayos críticos
utilizados en el proceso de fabricación.
3.2.2.4. Control de los excipientes
a) Se presentará una relación de todos los materiales necesarios
para fabricar el (los) excipiente(s), identificando cuando se emplea cada material
en el proceso. Se facilitará información sobre la calidad y el
control de dichos materiales. También se presentará información
que demuestre que los materiales cumplen los estándares apropiados para
su utilización prevista.
En todos los casos, los colorantes deberán reunir los requisitos que
se establecen en las Directivas 78/25/CEE y/o 94/36/CE. Además, los colorantes
deberán cumplir los criterios de pureza establecidos en virtud de la
Directiva 95/45/CE, modificada.
b) Deberán detallarse las especificaciones de cada excipiente, así
como su justificación. Se describirán y validarán debidamente
los procedimientos analíticos.
c) Se prestará atención específica a los excipientes de
origen humano o animal.
Respecto a las medidas específicas relativas a la prevención
de la transmisión de encefalopatías espongiformes animales, el
solicitante deberá demostrar asimismo que el medicamento está
fabricado con arreglo a la Nota Explicativa sobre Minimización del Riesgo
de Transmisión de los Agentes de la Encefalopatía Espongiforme
Animal a través de Medicamentos y sus actualizaciones, publicada por
la Comisión en el Diario Oficial de la Unión Europea.
Para demostrar el cumplimiento de lo dispuesto en la mencionada Nota Explicativa,
se podrá presentar, preferiblemente, un certificado de idoneidad con
la monografía relativa a Encefalopatías Espongiformes Transmisibles
de la Farmacopea Europea, o bien los datos científicos que corroboran
dicho cumplimiento.
d) Nuevos excipientes
Para los excipientes utilizados por primera vez en un medicamento o para una
nueva vía de administración, se presentarán con arreglo
al formato de la sustancia activa previamente descrito todos los datos de fabricación,
caracterización y controles, haciendo referencia cruzada a los datos
de apoyo relativos a seguridad, tanto clínicos como no clínicos.
Se presentará un documento en el que figurará la información
pormenorizada de carácter químico, farmacéutico y biológico.
Dicha información deberá presentarse en el mismo orden que el
capítulo dedicado al (los) principio(s) activo(s) del módulo 3.
La información relativa a nuevos excipientes podrá presentarse
como documento independiente según el formato descrito en los párrafos
anteriores. En caso de que el solicitante no sea el fabricante del nuevo excipiente,
el mencionado documento independiente deberá ponerse a disposición
del solicitante para su presentación a la autoridad competente.
En el módulo 4 del expediente se ofrecerá información suplementaria
sobre los estudios de toxicidad con el excipiente novedoso.
En el módulo 5 se presentarán los estudios clínicos.
3.2.2.5. Control del producto terminado
A efectos de control del producto terminado, se entenderá por lote de
un medicamento una entidad que comprenda todas las unidades de una forma farmacéutica
que provengan de una misma cantidad inicial de material y hayan sido sometidas
a la misma serie de operaciones de fabricación y esterilización
o, en el caso de un proceso de producción continuo, todas las unidades
fabricadas en un lapso de tiempo determinado.
Salvo debida justificación, la desviación máxima tolerable
del contenido del principio activo en el producto acabado no podrá ser
superior a +- 5 % en el momento de fabricación.
Se presentará información detallada sobre las especificaciones,
la justificación (liberación y período de validez) de su
elección, los métodos de análisis y su validación.
3.2.2.6. Estándares o materiales de referencia
Se determinarán y describirán detalladamente los Estándares
y materiales de referencia utilizados para poner a prueba el producto terminado,
en caso de que no se hayan presentado anteriormente en el apartado relativo
a la sustancia activa.
3.2.2.7. Envase y cierre del producto terminado
Se entregará la descripción del envase y el sistema o sistemas
de cierre, incluyendo la identidad de cada material de acondicionamiento primario
y sus especificaciones. En las especificaciones se incluirán la descripción
e identificación. Se incluirán, cuando proceda, los métodos
no recogidos en la farmacopea (con validación).
Para los materiales de acondicionamiento exterior no funcionales únicamente
se ofrecerá una breve descripción. Para los materiales de embalaje
exterior funcionales se ofrecerá información suplementaria.
3.2.2.8. Estabilidad del producto terminado
a) Deberán resumirse los tipos de estudios realizados, los protocolos
empleados y los resultados de los estudios.
b) Se presentarán con el formato adecuado los resultados pormenorizados
de los estudios sobre estabilidad, incluida la información relativa a
los procedimientos de análisis seguidos para obtener los datos, así
como la validación de dichos procedimientos; para las vacunas, se proporcionará
la información sobre la estabilidad acumulativa en aquellos casos en
que sea pertinente.
c) Se facilitarán el protocolo de estabilidad tras la aprobación
y el compromiso de estabilidad.
4.
MÓDULO 4: INFORMES NO CLíNICOS
4.1. Formato y presentación
El esquema general del módulo 4 es el siguiente:
- Índice
- Informes de estudios
- Farmacología,
- Farmacodinámica primaria
- Farmacodinámica secundaria
- Farmacología de seguridad
- Interacciones farmacodinámicas
- Farmacocinética
- Métodos analíticos e informes de validación
- Absorción
- Distribución
- Metabolismo
- Excreción
- Interacciones farmacocinéticas (no clínicas)
- Otros estudios de farmacocinética
- Toxicología
- Toxicidad por dosis única
- Toxicidad por administración continuada
- Genotoxicidad
- In vitro
- In vivo (incluidas las evaluaciones toxicocinéticas de apoyo)
- Carcinogénesis
- Estudios a largo plazo
- Estudios a corto o medio plazo
- Otros estudios
- Toxicidad en la reproducción y el desarrollo
- Fertilidad y desarrollo embrionario inicial
- Desarrollo embrionario y fetal
- Desarrollo prenatal y posnatal
- Estudios en los que se administran dosis a las crías (animales jóvenes)
y/o se evalúan posteriormente
- Tolerancia local
- Otros estudios sobre toxicidad
- Antigenicidad
- Inmunotoxicidad
- Estudios mecanicistas
- Dependencia
- Metabolitos
- Impurezas
- Otros
- Referencias bibliográficas.
4.2. Contenido: principios y requisitos básicos
Deberá prestarse particular atención a los elementos seleccionados
siguientes.
(1) Las pruebas toxicológicas y farmacológicas deberán
poner de manifiesto lo siguiente:
a) la toxicidad potencial del producto y los efectos peligrosos o no deseables
que pudieran producirse en seres humanos en las condiciones de uso propuestas,
valorándose estos efectos en función del proceso patológico
de que se trate;
b) sus propiedades farmacológicas, en relación a la posología
y la actividad farmacológica con el uso indicado en seres humanos. Todos
los resultados deberán ser fiables y de aplicación general. En
la medida en que sea conveniente, se utilizarán procedimientos matemáticos
y estadísticos para la elaboración de los métodos experimentales
y la valoración de los resultados.
Además, será necesario informar a los clínicos sobre el
potencial terapéutico y toxicológico del producto.
(2) En el caso de medicamentos biológicos tales como medicamentos inmunológicos
y medicamentos derivados de la sangre o el plasma humanos, puede ser necesario
adaptar los requisitos del presente módulo para algunos productos determinados;
por esta razón, el solicitante deberá justificar el programa de
las pruebas.
Al fijar el programa de las pruebas, deberán tenerse en cuenta los siguientes
puntos:
todas las pruebas que requieran la administración reiterada del producto
se diseñarán de modo que tengan en consideración la posible
inducción de anticuerpos e interferencia por parte de éstos;
deberá preverse un estudio de la función reproductora, de la toxicidad
embrionaria, fetal y perinatal, del potencial mutágeno así como
del potencial carcinógeno. Cuando los efectos sean atribuibles a componentes
distintos de la sustancia o sustancias activas, el estudio podrá sustituirse
por la validación de la eliminación de aquéllos.
(3) Se deberá investigar la toxicología y la farmacocinética
de un excipiente que se utilice por primera vez en el ámbito farmacéutico.
(4) Cuando se dé la posibilidad de una degradación significativa
durante el almacenamiento del medicamento, deberá tomarse en consideración
la toxicología de los productos de la degradación.
4.2.1. Farmacología
El estudio de farmacología deberá efectuarse siguiendo dos planteamientos
distintos.
- En primer lugar, las acciones relacionadas con el uso terapéutico
propuesto deberán estudiarse y describirse de manera adecuada. Siempre
que sea posible se realizarán ensayos reconocidos y validados, tanto
in vivo como in vitro. Deberán describirse técnicas experimentales
novedosas de manera suficientemente pormenorizada para que puedan reproducirse.
Los resultados se expresarán en términos cuantitativos, utilizando,
por ejemplo, curvas dosis-efecto y tiempo-efecto, etc. En la medida de lo posible,
se establecerán comparaciones con los datos correspondientes a una sustancia
o sustancias con una acción terapéutica análoga.
- En segundo lugar, el solicitante deberá investigar las posibles repercusiones
farmacodinámicas no deseadas de la sustancia en las funciones fisiológicas.
Tales investigaciones se realizarán en exposiciones correspondientes
a la gama terapéutica prevista y por encima de la misma. Las técnicas
experimentales, a no ser que sean las que se utilicen habitualmente, se describirán
de forma tal que permitan su reproducción, debiendo el investigador demostrar
su validez. Deberá estudiarse todo indicio de modificación de
las respuestas derivadas de la administración reiterada de la sustancia.
Respecto a la interacción farmacodinámica de los medicamentos,
las pruebas de combinaciones de principios activos podrán justificarse
bien por necesidades farmacológicas, bien por indicaciones clínicas.
En el primer caso, el estudio farmacodinámico deberá poner de
manifiesto aquellas interacciones que hagan recomendable la combinación
para el uso clínico. En el segundo caso, cuando la experimentación
clínica tenga por fin justificar científicamente la combinación
de sustancias, la investigación deberá determinar si los efectos
esperados de la combinación pueden demostrarse en animales y, como mínimo,
la importancia de las reacciones adversas.
4.2.2. Farmacocinética
Se entiende por farmacocinética el estudio del conjunto de procesos
que sufre el principio activo y sus metabolitos en el organismo. Comprende el
estudio de la absorción, la distribución, el metabolismo (biotransformación)
y la excreción de las sustancias.
El estudio de estas distintas fases se puede llevar a cabo principalmente con
métodos físicos, químicos o en su caso biológicos,
y mediante la observación de la actividad farmacodinámica real
de la propia sustancia.
Los datos referentes a la distribución y eliminación serán
necesarios en todos los casos en que dichos datos resulten indispensables para
determinar las dosis administrables a seres humanos, así como en las
sustancias quimioterapéuticas (antibióticos, etc.) y en las sustancias
cuyo uso se base en efectos no farmacodinámicos (por ejemplo, numerosos
agentes de diagnóstico, etc.).
También pueden realizarse estudios in vitro, con la ventaja de la utilización
de material humano para su comparación con material animal (es decir,
fijación con proteínas, metabolismo, interacción entre
medicamentos).
Es necesario el estudio farmacocinético de todas las sustancias farmacológicamente
activas. Cuando se trate de nuevas combinaciones de sustancias conocidas que
hayan sido estudiadas con arreglo a las disposiciones de la presente Directiva,
no será necesario exigir las investigaciones farmacocinéticas
si las pruebas de toxicidad y la experimentación clínica justifican
su omisión.
El programa farmacocinético se elaborará de modo que sean posibles
la comparación y la extrapolación entre animales y seres humanos.
4.2.3. Toxicología
a) Toxicidad por dosis única
Una prueba de toxicidad por dosis única es un estudio cualitativo y
cuantitativo de las reacciones tóxicas que pueden derivarse de una administración
única del principio o principios activos contenidos en el medicamento,
en las proporciones y en el estado físico-químico en que están
presentes en el producto.
La prueba de toxicidad por dosis única puede realizarse con arreglo a
las orientaciones pertinentes publicadas por la Agencia.
b) Toxicidad por administración continuada
Las pruebas de toxicidad por administración continuada tendrán
por objeto revelar las alteraciones funcionales y/o anatomo-patológicas
subsiguientes a la administración repetida del principio activo o de
la combinación de principios activos en cuestión y establecer
de qué modo se relacionan dichas alteraciones con la posología.
Generalmente es aconsejable realizar dos pruebas: una a corto plazo, durante
dos a cuatro semanas, y la otra a largo plazo. La duración de la segunda
prueba dependerá de las condiciones de la utilización clínica.
Su objeto es describir los posibles efectos nocivos, a los que deberá
prestarse atención en los estudios clínicos. La duración
se define en las directrices correspondientes publicadas por la Agencia.
c) Genotoxicidad
El objeto del estudio del potencial mutagénico y clastogénico
es revelar las alteraciones que puede causar una sustancia en el material genético
de las personas y las células. Las sustancias mutagénicas pueden
representar un riesgo para la salud, ya que la exposición a un mutágeno
supone el riesgo de inducir una mutación germinal, con la posibilidad
de trastornos hereditarios, y el riesgo de mutaciones somáticas, que
incluso pueden ser causa de cáncer Dicho estudio será obligatorio
para cualquier sustancia nueva.
d) Carcinogénesis
Se exigirá habitualmente efectuar pruebas dirigidas a revelar efectos
carcinógenos:
1. Estos estudios se realizarán con todos los medicamentos cuya utilización
clínica se prevea para un período prolongado de la vida del paciente,
bien de manera continuada, bien de manera reiterada e intermitente.
2. Los estudios relativos a determinados medicamentos se recomiendan si se piensa
que representan un potencial carcinogenético, por ejemplo tomando como
referencia un producto de la misma clase o estructura, o a raíz de pruebas
obtenidas en estudios de toxicidad por administración continuada.
3. No son necesarios los estudios con componentes inequívocamente genotóxicos,
ya que se supone que son carcinógenos que afectan a distintas especies
y suponen un riesgo para el ser humano. Si se pretende administrar un medicamento
de este tipo de manera crónica a seres humanos, puede resultar necesario
un estudio crónico para detectar efectos tumorigénicos precoces.
e) Toxicidad en la reproducción y el desarrollo
La investigación acerca de posibles alteraciones de la función
reproductora masculina o femenina, así como los efectos nocivos para
los descendientes, deberá realizarse mediante las pruebas pertinentes.
En ellas se incluyen los estudios sobre la repercusión en la función
reproductora masculina y femenina, sobre los efectos tóxicos y teratógenos
en todas las fases de desarrollo desde la concepción a la madurez sexual,
así como los efectos latentes, cuando el medicamento investigado ha sido
administrado a la mujer durante el embarazo.
Deberá justificarse de manera adecuada la omisión de tales pruebas.
En función de la utilización indicada del medicamento, podrá
justificarse la realización de estudios suplementarios acerca del desarrollo
de la descendencia cuando se administra el medicamento.
Los estudios de toxicidad embrionaria y fetal se realizarán normalmente
con dos especies de mamíferos, una de las cuales deberá no ser
un roedor. Los estudios perinatales y posnatales se llevarán a cabo con
al menos una especie. Si se sabe que el metabolismo de un medicamento en determinada
especie es similar al del hombre, es deseable incluir esa especie. También
es deseable que una de las especies sea la misma que la de los estudios de toxicidad
por administración continuada.
La concepción del estudio se determinará teniendo en cuenta el
estado de los conocimientos científicos en el momento de presentarse
la solicitud.
f) Tolerancia local
El objetivo de los estudios de tolerancia local es determinar si los medicamentos
(tanto los principios activos como los excipientes) se toleran en los lugares
del cuerpo que pueden entrar en contacto con el medicamento como consecuencia
de su administración durante el uso clínico. El procedimiento
de prueba debe ser tal que todo efecto mecánico de la administración,
o las acciones puramente fisicoquímicas del producto, puedan distinguirse
de los efectos toxicológicos o farmacodinámicos.
Deberán realizarse las pruebas sobre tolerancia local con el preparado
que se está desarrollando para su uso humano, utilizando el vehículo
y/o los excipientes en el tratamiento del grupo o grupos de control. Si es preciso,
se incluirán controles y sustancias de referencia positivos.
La concepción de las pruebas de tolerancia local (elección de
la especie, duración, frecuencia y vía de administración,
dosificación) dependerá del problema que deba investigarse y las
condiciones propuestas de administración en la utilización clínica.
Deberá realizarse la reversibilidad de las lesiones locales cuando resulte
pertinente.
Los estudios en animales podrán sustituirse por pruebas validadas in
vitro, siempre que los resultados de las pruebas sean de calidad y utilidad
análogas para los fines de la evaluación de la seguridad.
En el caso de las sustancias químicas aplicadas a la piel (por ejemplo,
dérmicas, rectales, vaginales) se evaluará el potencial de sensibilización
como mínimo en uno de los sistemas de prueba actualmente disponibles
(ensayo con cobayas o ensayo de ganglio linfático local).
5.
MÓDULO 5: INFORMES DE ESTUDIOS CLíNICOS
5.1. Formato y presentación
El esquema general del módulo 5 es el siguiente:
- Índice de informes de estudios clínicos
- Listado en forma de tabla de todos los estudios clínicos
- Informes de los estudios clínicos
- Informes de estudios biofarmacéuticos
- Informes de estudios de biodisponibilidad
- Informes de estudios comparativos de biodisponibilidad y bioequivalencia
- Informes de estudios de correlación in vitro - in vivo
- Informes de métodos bioanalíticos y analíticos
- Informes de estudios sobre farmacocinética mediante biomateriales humanos
- Informes de estudios de fijación con proteínas del plasma
- Informes de estudios sobre metabolismo hepático e interacción
- Informes de estudios mediante otros biomateriales humanos
- Informes de estudios de farmacocinética humana
- Informes de estudios de farmacocinética y tolerancia inicial en sujetos
sanos
- Informes de estudios de farmacocinética y tolerancia inicial en pacientes
- Informes de estudios de farmacocinética de factores intrínsecos
- Informes de estudios de farmacocinética de factores extrínsecos
- Informes de estudios de farmacocinética en la población
- Informes de estudios de farmacodinámica humana
- Informes de estudios de farmacodinámica y farmacocinética/farmacodinámica
en sujetos sanos
- Informes de estudios de farmacodinámica y farmacocinética/farmacodinámica
en pacientes
- Informes de estudios sobre eficacia y seguridad
- Informes de estudios clínicos controlados relativos a la indicación
declarada
- Informes de estudios clínicos no controlados
- Informes de análisis de datos procedentes de diversos estudios, incluido
cualquier meta-análisis, análisis comparativo (bridging analyses)
y análisis integrado formal.
- Otros informes de estudio
- Informes de experiencia posterior a la comercialización
- Referencias bibliográficas.
5.2. Contenido: principios y requisitos básicos
Deberá prestarse particular atención a los elementos seleccionados
siguientes.
a) Los datos clínicos que se suministren en cumplimiento de lo dispuesto
en la letra i) del apartado 3 del artículo 8 y en el apartado 1 del artículo
10 deberán permitir formarse una opinión suficientemente fundada
y científicamente válida acerca de si la especialidad responde
a los criterios previstos para la concesión de la autorización
de comercialización. Por este motivo, es preceptivo que se comuniquen
los resultados de todos los ensayos clínicos que se hayan realizado,
tanto favorables como desfavorables.
b) Los ensayos clínicos deberán ir siempre precedidos de las
necesarias pruebas farmacológicas y toxicológicas en animales,
efectuadas con arreglo a lo dispuesto en el módulo 4 del presente anexo.
El investigador deberá conocer las conclusiones de los exámenes
farmacológico y toxicológico y, por tanto, el solicitante deberá
proporcionarle, como mínimo, el manual del investigador, que consistirá
en toda la información pertinente conocida antes del inicio de un ensayo
clínico, e incluirá datos químicos, farmacéuticos
y biológicos, datos toxicológicos, farmacocinéticos y farmacodinámicos
en animales, así como los resultados de ensayos clínicos anteriores,
con datos útiles que justifiquen la naturaleza, la escala y la duración
del ensayo propuesto; a petición del investigador se deberán suministrar
los informes farmacológicos y toxicológicos completos. Cuando
se trate de materias de origen humano o animal, se emplearán todos los
medios disponibles antes del inicio del ensayo para garantizar que no se transmiten
agentes infecciosos.
c) Los titulares de la autorización de comercialización deberán
tomar las medidas necesarias para que los documentos de los ensayos clínicos
esenciales (incluidos los impresos de recogida de datos) distintos del expediente
médico del sujeto sean custodiados por los propietarios de los datos:
- durante un mínimo de 15 años tras la finalización o interrupción
del ensayo, o
- durante un mínimo de dos años tras la concesión de la
última autorización de comercialización en la Comunidad
Europea y en aquellos casos en que no haya solicitudes de comercialización
pendientes o previstas en la Comunidad Europea, o
- durante un mínimo de dos años tras la interrupción oficial
del desarrollo clínico del producto objeto de investigación.
El expediente médico del sujeto deberá ser custodiado con arreglo
a la normativa aplicable y conforme al período máximo permitido
por el hospital, institución o consulta privada.
No obstante, podrán retenerse los documentos durante un período
más largo, si así lo exigen las disposiciones normativas aplicables
o el acuerdo con el promotor. Corresponderá al promotor informar al hospital,
institución o consulta privada acerca del momento en que no será
preciso continuar conservando dichos documentos.
El promotor o el propietario de los datos conservará toda la restante
documentación relativa al ensayo durante el período de validez
del medicamento. Entre dicha documentación deberán figurar: el
protocolo, incluidos la justificación, los objetivos, el diseño
estadístico y la metodología del ensayo, con las condiciones en
las que se efectúe y gestione, así como los pormenores del medicamento
de investigación, el medicamento de referencia y/o el placebo que se
empleen; los procedimientos normalizados de trabajo; todos los informes escritos
sobre el protocolo y los procedimientos; el manual del investigador; el cuaderno
de recogida de datos de cada sujeto; el informe final; el (los) certificado(s)
de auditoría, cuando se disponga de él (ellos). El promotor o
el propietario subsiguiente conservará el informe final hasta pasados
cinco años tras haberse agotado el plazo de validez del medicamento.
Además de los ensayos que se realicen dentro de la Comunidad Europea,
el titular de la autorización de comercialización tomará
todas las medidas suplementarias para el archivo de la documentación
con arreglo a lo dispuesto en la Directiva 2001/20/CE y en las directrices detalladas
de aplicación.
Deberá documentarse todo cambio que se produzca en la propiedad de los
datos.
Todos los datos y documentos deberán ponerse a disposición de
las autoridades competentes, si éstas así lo solicitan.
d) Los datos sobre cada ensayo clínico deberán estar suficientemente
detallados para permitir un juicio objetivo, y contendrán, en particular:
- el protocolo, incluyendo la justificación, los objetivos, el diseño
estadístico y la metodología del ensayo, con las condiciones en
las que se efectúa y gestiona, así como los pormenores del medicamento
objeto de estudio que se emplee;
- el (los) certificado(s) de auditoría, cuando se disponga de él
(ellos);
- la lista de investigadores; cada investigador deberá indicar su nombre,
domicilio, cargo, titulación y obligaciones clínicas, hacer constar
dónde se llevó a cabo el ensayo y reunir la información
relativa a cada uno de los pacientes, incluyendo los impresos de recogida de
datos de cada sujeto;
- el informe final, firmado por el investigador y para ensayos multicéntricos
por todos los investigadores o por el investigador responsable de la coordinación.
e) Los anteriores datos sobre los ensayos clínicos se remitirán
a las autoridades competentes. No obstante, el solicitante podrá omitir
parte de esta información con el consentimiento de dichas autoridades.
A petición de éstas, deberá enviar sin demora la documentación
completa.
El investigador deberá pronunciarse, en sus conclusiones de la experimentación,
sobre la seguridad del producto en las condiciones normales de utilización,
su tolerancia y su eficacia, aportando todas las precisiones que resulten útiles
sobre las indicaciones y contraindicaciones, la posología y la duración
media del tratamiento, así como, en caso necesario, las precauciones
particulares de uso y los signos clínicos de sobre dosificación.
Cuando informe sobre los resultados de un estudio multicéntrico, el investigador
principal deberá expresar, en sus conclusiones, su opinión sobre
la seguridad y eficacia del medicamento que es objeto del estudio en nombre
de todos los centros.
f) Se resumirán las observaciones clínicas de cada ensayo, indicando:
1) el número de los sujetos tratados, distribuidos por sexo;
2) la selección y la distribución por edad de los grupos de pacientes
que son objeto de investigación y las pruebas comparativas;
3) el número de pacientes que hayan sido retirados prematuramente de
los ensayos, así como los motivos para ello;
4) en caso de que se hayan llevado a cabo ensayos controlados según lo
dispuesto anteriormente, si el grupo control:
- no ha sido sometido a tratamiento,
- ha recibido un placebo,
- ha recibido otro medicamento de efecto conocido,
- ha recibido un tratamiento no medicamentoso;
5) la frecuencia de las reacciones adversas observadas;
6) todas las precisiones sobre los pacientes que presenten una especial sensibilidad
(ancianos, niños, mujeres embarazadas o en período de menstruación)
o cuyo estado fisiológico o patológico exija una especial consideración;
7) parámetros o criterios para evaluar la eficacia, así como los
resultados referentes a estos parámetros;
8) una evaluación estadística de los resultados, en la medida
en que se requiera por el diseño de los ensayos y las variables implicadas.
g) Además, el investigador deberá en todo caso señalar
sus observaciones sobre:
1) todo indicio de habituación, adicción o dificultad en pacientes
que dejan de tomar el medicamento;
2) las interacciones observadas con otros medicamentos que se administren simultáneamente;
3) los criterios con arreglo a los cuales se excluyó a determinados pacientes
de los ensayos;
4) toda muerte que se haya producido durante el ensayo o durante el período
de seguimiento.
h) Los datos relativos a una nueva combinación de sustancias medicamentosas
deberán ser idénticos a los que se exigen en el caso de medicamentos
nuevos, y deberán justificar la seguridad y la eficacia de la combinación.
i) Será obligatorio justificar la ausencia parcial o total de datos.
Si se producen resultados imprevistos a lo largo de los ensayos, deberán
realizarse y documentarse ensayos preclínicos, toxicológicos y
farmacológicos adicionales.
j) Si el medicamento está destinado a ser administrado de forma prolongada,
habrá que suministrar datos sobre toda modificación de la acción
farmacológica tras una administración reiterada, así como
sobre la determinación de una dosificación a largo plazo.
5.2.1. Informes de estudios biofarmacéuticos
Deberán presentarse informes de estudios de biodisponibilidad, biodisponibilidad
comparativa, bioequivalencia, correlación in vitro - in vivo y métodos
bioanalíticos y analíticos.
Además, deberá evaluarse la biodisponibilidad cuando sea necesario
para demostrar la bioequivalencia de los medicamentos a los que se refiere la
letra a) del apartado 1 del artículo 10.
5.2.2. Informes de estudios sobre farmacocinética mediante biomateriales
humanos
A efectos del presente anexo, se entenderá por biomateriales humanos
todas las proteínas, células, tejidos y materiales conexos de
origen humano que se utilizan in vivo o ex vivo para evaluar las propiedades
farmacocinéticas de las sustancias medicamentosas.
A este respecto, se entregarán informes de estudios de fijación
con proteínas del plasma, estudios de metabolismo hepático e interacción
de sustancias activas y estudios que utilicen otros biomateriales humanos.
5.2.3. Informes de estudios de farmacocinética humana
a) Se describirán las siguientes características farmacocinéticas:
- absorción (velocidad y magnitud),
- distribución,
- metabolismo,
- excreción.
Deberán describirse los aspectos significativos desde el punto de vista
clínico, incluyendo la implicación de los datos cinéticos
para el régimen de dosificación, especialmente para los pacientes
de riesgo, y las diferencias entre el hombre y las especies animales utilizadas
en los estudios preclínicos.
Además de los estudios normales de farmacocinética de muestras
múltiples, los análisis farmacocinéticos de la población
basados en un muestreo disperso durante los estudios clínicos también
pueden abordar las cuestiones relativas a la contribución de los factores
intrínsecos y extrínsecos a la variabilidad de la relación
dosis-respuesta farmacocinética. Se entregarán informes de estudios
de farmacocinética y tolerancia inicial en sujetos sanos y en pacientes,
informes de estudios farmacocinéticos destinados a evaluar la repercusión
de los factores intrínsecos y extrínsecos e informes de estudios
farmacocinéticos de la población.
b) Cuando el medicamento vaya a administrarse, de forma habitual, simultáneamente
con otros medicamentos, deberán proporcionarse datos sobre las pruebas
de administración conjunta realizadas para demostrar posibles modificaciones
de la acción farmacológica.
Se investigarán las interacciones farmacocinéticas entre los principios
activos y otros medicamentos o sustancias.
5.2.4. Informes de estudios de farmacodinámica humana
a) Deberá demostrarse la acción farmacodinámica correlacionada
con la eficacia, incluyendo:
- la relación dosis-respuesta y su curso temporal,
- la justificación de la posología y las condiciones de administración,
- cuando sea posible, el modo de acción.
Se describirá la acción farmacodinámica no relacionada
con la eficacia.
La demostración de efectos farmacodinámicos en seres humanos no
bastará por sí misma para establecer conclusiones en cuanto a
un posible efecto terapéutico.
b) Cuando el medicamento vaya a administrarse, de forma habitual, simultáneamente
con otros medicamentos, deberán proporcionarse datos sobre las pruebas
de administración conjunta realizadas para demostrar posibles modificaciones
de la acción farmacológica.
Se investigarán las interacciones farmacodinámicas entre las sustancias
activas y otros medicamentos o sustancias.
5.2.5. Informes de estudios sobre eficacia y seguridad
5.2.5.1. Informes de estudios clínicos controlados relativos a la
indicación declarada
En general, los ensayos clínicos se efectuarán en forma de ensayos
clínicos controlados siempre que sea posible, aleatorizados y, según
convenga, en comparación con un placebo y un medicamento conocido, cuyo
valor terapéutico esté bien establecido; cualquier otro diseño
deberá justificarse. El tratamiento asignado al grupo control variará
según los casos y dependerá también de consideraciones
éticas y del ámbito terapéutico. En este sentido, en ocasiones
puede resultar más conveniente comparar la eficacia de un nuevo medicamento
con el efecto de un medicamento conocido, cuyo valor terapéutico esté
bien establecido, y no con el efecto de un placebo.
(1) En la medida de lo posible, y muy especialmente en ensayos en los que el
efecto del producto no pueda medirse objetivamente, se tomarán medidas
para evitar un sesgo, incluyendo métodos de aleatorización y métodos
ciegos (de doble ciego).
(2) El protocolo del ensayo deberá incluir una descripción pormenorizada
de los métodos estadísticos a los que se recurra, del número
de pacientes y las razones por las que se incluyen (con el cálculo del
valor estadístico del ensayo), el nivel de significación que se
use y una descripción de la unidad estadística. Deben documentarse
las medidas que se adopten para evitar el sesgo, en particular los métodos
de aleatorización. La inclusión de un gran número de pacientes
a lo largo de un ensayo no deberá considerarse en ningún caso
el sustituto válido de un ensayo controlado bien ejecutado.
Los datos sobre seguridad deberán examinarse teniendo en cuenta las directrices
publicadas por la Comisión, prestando especial atención a hechos
que den como resultado la alteración de la dosis o la necesidad de medicación
concomitante, hechos nocivos graves, hechos que provoquen la retirada y fallecimientos.
Deberán determinarse todos los pacientes o grupos que corren mayor riesgo
y se prestará especial atención a los pacientes potencialmente
vulnerables que puedan resultar poco numerosos, por ejemplo, niños, embarazadas,
personas de edad avanzada delicadas, personas con fuertes anomalías de
metabolismo o excreción, etc. Se describirá la repercusión
de la evaluación de la seguridad para los posibles empleos del medicamento.
5.2.5.2. Informes de estudios clínicos no controlados, informes de
análisis de datos obtenidos en diversos estudios y otros informes de
estudios clínicos
Deberán facilitarse todos estos informes.
5.2.6. Informes de experiencia posterior a la comercialización
Si el medicamento ya está autorizado en terceros países, deberá
proporcionarse información sobre reacciones adversas al medicamento en
cuestión y a medicamentos que contengan los mismos principios activos,
a ser posible en relación con la tasa de utilización.
5.2.7. Cuadernos de recogida de datos y listados de pacientes
Al presentar los cuadernos de recogida de datos y las listas de pacientes con
arreglo a las directrices pertinentes publicadas por la Agencia, deberán
facilitarse y presentarse en el mismo orden que los informes de estudios clínicos
e indexarse por estudio.
PARTE II
EXPEDIENTES DE AUTORIZACIÓN DE COMERCIALIZACIÓN Y REQUISITOS ESPECÍFICOS
Algunos medicamentos presentan características específicas que
hacen necesaria la adaptación de todos los requisitos del expediente
de solicitud de autorización de comercialización que se establecen
en la parte I del presente anexo. Con el fin de tener en cuenta estas situaciones
especiales, los solicitantes utilizarán una presentación adaptada
y adecuada del expediente.
1. SOLICITUDES BIBLIOGRAFICAS
Se aplicarán las normas específicas que se exponen a continuación
a los medicamentos cuyo(s) principio(s) activo(s) tengan, tal como se menciona
en el inciso ii) de la letra a) del apartado 1 del artículo 10, un "uso
medicinal claramente establecido" (o suficientemente comprobado), con una
eficacia reconocida y un nivel aceptable de seguridad.
El solicitante deberá presentar los módulos 1, 2 y 3 tal como
se describen en la parte I del presente anexo.
Para los módulos 4 y 5, deberán abordarse las características
clínicas y no clínicas mediante una bibliografía científica
detallada.
Las siguientes normas específicas serán de aplicación para
demostrar la existencia de un uso médico suficientemente comprobado:
a) Los factores que han de tenerse en cuenta para determinar un uso médico
suficientemente comprobado de los componentes del medicamento son los siguientes:
- el período durante el que se ha utilizado una sustancia,
- los aspectos cuantitativos del empleo de la misma,
- el grado de interés científico de su utilización (que
se refleja en la bibliografía científica publicada),
- la coherencia de las evaluaciones científicas.
Por tanto, pueden ser necesarios períodos de tiempo diferentes a fin
de establecer el uso médico suficientemente comprobado de las diferentes
sustancias. En todo caso, el período de tiempo necesario para establecer
que un componente de un medicamento tiene un uso medicinal suficientemente comprobado
no podrá ser inferior a diez años, contados a partir de la primera
utilización sistemática y documentada de esa sustancia como medicamento
dentro de de la Comunidad.
b) La documentación presentada por el solicitante deberá cubrir
todos los aspectos de la evaluación de la seguridad y/o de la eficacia
e incluir o hacer referencia a un estudio bibliográfico pertinente, que
tenga en cuenta los estudios previos y posteriores a la comercialización
y la literatura científica publicada relativa a la experimentación
en forma de estudios epidemiológicos y, en particular, de estudios epidemiológicos
comparativos. Deberán comunicarse todos los documentos existentes, tanto
favorables como desfavorables. Respecto a las disposiciones sobre "uso
médico suficientemente comprobado", es particularmente necesario
aclarar que la "referencia bibliográfica" a otras pruebas (estudios
posteriores a la comercialización, estudios epidemiológicos, etc.),
y no sólo los datos relacionados con ensayos, puede servir como prueba
válida de la seguridad y eficacia de un medicamento si una solicitud
explica y justifica satisfactoriamente la utilización de estas fuentes
de información.
c) Se prestará atención particular a cualquier informacion omitida
y se justificará por qué puede afirmarse la existencia de un nivel
aceptable de seguridad y/o eficacia, pese a la ausencia de determinados estudios.
d) En las visión general de las partes no clínicas y/o clínicas
deberá explicarse la relevancia de todos los datos presentados relativos
a un producto diferente de aquel que será comercializado. Se deberá
valorar si el producto examinado puede considerarse similar al producto cuya
autorización de comercialización se ha solicitado a pesar de las
diferencias existentes.
e) La experiencia posterior a la comercialización de otros productos
que contengan los mismos componentes revestirá particular importancia
y los solicitantes deberán insistir especialmente en este aspecto.
2. MEDICAMENTOS ESENCIALMENTE SIMILARES
a) Las solicitudes basadas en el inciso i) de la letra a) del apartado 1 del
artículo 10 (medicamentos esencialmente similares) deberán contener
los datos descritos en los módulos 1, 2 y 3 de la parte I del presente
anexo, siempre que el solicitante haya obtenido el consentimiento del titular
de la autorización de comercialización original para hacer referencia
cruzada al contenido de sus módulos 4 y 5.
b) Las solicitudes basadas en el inciso iii) de la letra a) del apartado 1
del artículo 10 (medicamentos esencialmente similares, esto es, genéricos)
incluirán los datos descritos en los módulos 1, 2 y 3 de la parte
I del presente anexo, junto con los datos que demuestren la biodisponibilidad
y la bioequivalencia con el medicamento original, siempre que este no sea un
medicamento biológico (véase el punto 4 de la parte II, Medicamentos
biológicos similares).
Los resúmenes visiones generales no clínicas/clínicas de
dichos productos se centrarán especialmente en los siguientes elementos:
- los motivos por los que se reclamaa la similaridad esencial;
- un resumen de las impurezas presentes en lotes del (los) principio(s) activo(s)
así como las del producto terminado (y, cuando proceda, los productos
de descomposición que se forman durante el almacenamiento) tal como se
propone paraser comercializador, acompañado de una evaluación
de dichas impurezas;
- una evaluación de los estudios de bioequivalencia o una justificación
por no haber realizado los estudios siguiendo las directrices sobre la "Investigación
de la biodisponibilidad y bioequivalencia";
- una actualización de la bibliografía publicada sobre la sustancia
y la presente solicitud; se aceptarán las anotaciones con este fin de
artículos de publicaciones especializadas;
- cada afirmación que figure en el resumen de las características
del producto no conocida o deducida a partir de las propiedades del medicamento
y/o su grupo terapéutico deberá discutirse en las resúmenes/visiones
generales de las partes no clínicas/clínicas y justificarse mediante
la bibliografía publicada o estudios suplementarios;
- si procede, el solicitante deberá aportar datos adicionales para probar
la equivalencia de las propiedades de seguridad y eficacia de las diferentes
sales, ésteres o derivados de un principio activo autorizado, en caso
de que reclame la similaridad esencial.
3. DATOS SUPLEMENTARIOS EXIGIDOS EN SITUACIONES ESPECÍFICAS
Cuando el principio activo de un medicamento esencialmente similar contenga
la misma fracción terapéutica que el medicamento autorizado original,
asociada a un complejo/derivado de sales/ésteres diferentes, habrá
de probarse que no se produce alteración alguna de la farmacocinética
de la fracción, la farmacodinamia y/o la toxicidad que pueda modificar
su perfil de seguridad/eficacia. De lo contrario, se considerará que
tal asociación constituye un nuevo principio activo.
En los casos en que el medicamento esté destinado a un uso terapéutico
diferente o se presente en una forma farmacéutica distinta o deba administrarse
por vías diferentes o con dosificación diferente, deberán
suministrarse los resultados de las pruebas toxicológicas y farmacológicas
apropiadas y/o de los ensayos clínicos.
4. MEDICAMENTOS BIOLÓGICOS SIMILARES
En el caso de los medicamentos biológicos lo dispuesto en el inciso
iii) de la letra a) del apartado 1 del artículo 10 puede no ser suficiente.
Si la información exigida en el caso de medicamentos esencialmente similares
(genéricos) no permite la demostración de la naturaleza análoga
de dos medicamentos biológicos, se deberán facilitar datos suplementarios,
en particular el perfil toxicológico y clínico.
Cuando un solicitante independiente solicite, una vez concluido el período
de protección de datos, una autorización de comercialización
de un medicamento biológico definido en el punto 3.2 de la parte I del
presente anexo que se relacione con un medicamento original que haya obtenido
la autorización de comercialización en la Comunidad, se aplicará
el enfoque que se expone a continuación.
- La información que habrá de facilitarse no se limitará
a los módulos 1, 2 y 3 (datos farmacéuticos, químicos y
biológicos), complementada con los datos sobre bioequivalencia y biodisponibilidad.
El tipo y la cantidad de datos suplementarios (esto es, datos toxicológicos,
datos no clínicos y datos clínicos pertinentes) se determinará
en cada caso, conforme a todas las directrices científicas pertinentes.
- Debido a la diversidad de medicamentos biológicos, la autoridad competente
determinará si es necesario exigir los estudios identificados previstos
en los módulos 4 y 5, teniendo en cuenta las características especiales
de cada medicamento.
Los principios generales que han de aplicarse se recogen en las directrices
publicadas por la Agencia, en las que se tienen en cuenta las características
de los medicamentos biológicos en cuestión. En caso de que el
medicamento autorizado originalmente tenga más de una indicación,
deberán justificarse la eficacia y la seguridad del medicamento que se
afirma es similar o, si es necesario, deberán demostrarse por separado
respecto a cada una de las indicaciones declaradas.
5. MEDICAMENTOS DE COMBINACIÓN FIJA
Las solicitudes basadas en la letra b) del apartado 1 del artículo
10 se referirán a medicamentos nuevos compuestos por dos principios activos
como mínimo que no han sido autorizadas anteriormente como medicamento
de combinación fija.
En el caso de esas solicitudes se presentará un expediente completo (módulos
1 a 5) para el medicamento de combinación fija. Si procede, se facilitará
información relativa a los lugares de fabricación y la evaluación
de la seguridad de los agentes extraños/externos.
6. DOCUMENTACIÓN PARA LAS SOLICITUDES DE AUTORIZACIÓN EN CIRCUNSTANCIAS
EXCEPCIONALES
Cuando, como se establece en el artículo 22, el solicitante pueda demostrar
que no puede suministrar datos completos sobre la eficacia y seguridad en las
condiciones normales de uso del producto, por alguna de las razones siguientes:
- los casos para los que están indicados los productos en cuestión
se presentan tan raramente que el solicitante no puede razonablemente estar
obligado a proporcionar las evidencias detalladas;
- el estado actual de desarrollo de la ciencia no permite proporcionar información
completa;
- principios de deontología médica comúnmente admitidos
prohíben recoger esta información,
podrá concederse la autorización de comercialización en
función de determinadas obligaciones específicas.
Entre dichas obligaciones podrán figurar las siguientes:
- el solicitante concluirá, dentro de un plazo especificado por la autoridad
competente, un programa de estudios determinado cuyos resultados constituirán
la base de una nueva evaluación de la relación riesgo/beneficio;
- la especialidad de que se trate sólo se expedirá con receta
médica y, en caso necesario, sólo se autorizará su administración
si se efectúa bajo estricto control médico, a ser posible en un
centro hospitalario y, cuando se trate de un radiofármaco, por parte
de una persona autorizada;
- el prospecto y cualquier otra información médica destacaráque,
en relación con determinados aspectos, no existen aún datos fiables
sobre el medicamento en cuestión.
7. SOLICITUDES MIXTAS DE AUTORIZACIÓN DE COMERCIALIZACIÓN
Por solicitudes mixtas de autorización de comercialización se
entenderán los expedientes de solicitud en los que los módulos
4 y/o 5 constan de una combinación de informes de estudios limitados
no clínicos y/o clínicos realizados por el solicitante y de referencias
bibliográficas. Todos los demás módulos se ajustarán
a la estructura descrita en la parte I del presente anexo. La autoridad competente
deberá aceptar el formato propuesto que presente el solicitante considerando
individualmente cada caso.
PARTE III
MEDICAMENTOS PARTICULARES
En esta parte se establecen los requisitos relacionados con la naturaleza de
determinados medicamentos.
1. MEDICAMENTOS BIOLÓGICOS
1.1. Medicamentos derivados del plasma
Respecto a los medicamentos derivados de sangre humana o plasma y no obstante
lo dispuesto en el módulo 3, los requisitos de los expedientes mencionados
en "información sobre los materiales de partida y materias primas"
referentes a los materiales de partida derivados de sangre o plasma humanos
podrán ser sustituidos por un Archivo Principal sobre Plasma Certificado
con arreglo a lo expuesto en la presente parte.
a) Principios
A efectos del presente anexo:
- Se entenderá por "Archivo Principal sobre Plasma" aquella
documentación independiente y separada del expediente de autorización
de comercialización que contenga toda la información pormenorizada
pertinente sobre las características de todo el plasma humano empleado
como material de partida y/o materia prima para la fabricación de subfracciones
o fracciones, componentes del excipiente y principio(s) activo(s), que forman
parte de los medicamentos o productos sanitarios mencionados en la Directiva
2000/70/CE del Parlamento Europeo y del Consejo, de 16 de noviembre de 2000,
que modifica la Directiva 93/42/CEE del Consejo en lo referente a los productos
sanitarios que incorporen derivados estables de la sangre o plasma humanos(11).
- Cada centro o establecimiento de fraccionamiento/tratamiento de plasma humano
deberá preparar y mantener al día el conjunto de información
pormenorizada pertinente a la que se hace referencia en el archivo principal
sobre plasma.
- El solicitante de una autorización de comercialización o el
titular de la autorización de comercialización presentará
el archivo principal sobre plasma a la Agencia o a la autoridad competente.
En caso de que el solicitante de una autorización de comercialización
o el titular de la misma no sean el titular del archivo principal sobre plasma,
este archivo deberá ponerse a disposición del solicitante o del
titular de la autorización de comercialización para su presentación
a la autoridad competente. En cualquier caso, el solicitante o titular de la
autorización de comercialización asumirá la responsabilidad
del medicamento.
- La autoridad competente que evalúa la autorización de comercialización
esperará a que la Agencia expida el certificado antes de tomar una decisión
sobre la solicitud.
- Todos los expedientes de autorización de comercialización que
contengan algún componente derivado de plasma humano deberán remitir
al archivo principal sobre plasma correspondiente al plasma utilizado como material
de partida o materia prima.
b) Contenido
Con arreglo a lo dispuesto en el artículo 109, modificado por la Directiva
2002/98/CE, referente a los requisitos que deben reunir los donantes y a la
verificación de las donaciones, el archivo principal sobre plasma incluirá
información sobre el plasma utilizado como material de partida o materia
prima, en concreto:
(1) Origen del plasma
(i) Información acerca de los centros o establecimientos en los que se
recoja la sangre o plasma, incluidas la inspección y aprobación,
y datos epidemiológicos sobre infecciones transmisibles por la sangre.
(ii) Centros o establecimientos de información en los que se realizan
análisis de las donaciones y bancos de plasma, incluida la categoría
de la inspección y aprobación.
(iii) Criterios de selección/exclusión de los donantes de sangre
y plasma.
(iv) Sistema implantado que permite rastrear el itinerario de cada donación
desde el establecimiento de recogida de sangre y plasma hasta los productos
terminados y viceversa.
(2) Calidad y seguridad del plasma
(i) Cumplimiento de las monografías de la Farmacopea europea.
(ii) Realización de análisis de las donaciones y bancos de sangre
y plasma para detectar agentes infecciosos, incluida la información sobre
los métodos de análisis y, en el caso de los bancos de plasma,
datos de validación acerca de los métodos de análisis empleados.
(iii) Características técnicas de las bolsas de recogida de sangre
y plasma, incluidos los datos sobre las soluciones anticoagulantes empleadas.
(iv) Condiciones de almacenamiento y transporte de plasma.
(v) Procedimientos para el mantenimiento de inventarios y/o períodos
de cuarentena.
(vi) Caracterización de la mezcla de plasma original.
(3) Sistema en funcionamiento entre el fabricante de medicamentos derivados
de plasma y/o la entidad que se ocupa del fraccionamiento o tratamiento del
plasma, por un parte, y los centros o establecimientos de recogida y ensayo
de la sangre y plasma, por otra, que define las condiciones de su interacción
y las especificaciones acordadas entre ellos.
Además, en el archivo principal sobre plasma se ofrecerá una lista
de los medicamentos para los que es válido el archivo, tanto los medicamentos
que han obtenido una autorización de comercialización como los
que están en vías de obtenerla, incluidos los medicamentos mencionados
en el artículo 2 de la Directiva 2001/20/CE del Parlamento Europeo y
del Consejo, relativa a la aproximación de las disposiciones legales,
reglamentarias y administrativas de los Estados miembros sobre la aplicación
de buenas prácticas clínicas en la realización de ensayos
clínicos de medicamentos de uso humano.
c) Evaluación y certificación
- En el caso de los medicamentos aún no autorizados, el solicitante
de la autorización de comercialización presentará un expediente
completo a la autoridad competente, que deberá ir acompañado por
un archivo principal sobre plasma aparte en caso de que éste no exista
ya.
- El archivo principal sobre plasma estará sujeto a una evaluación
científica y técnica que realiza la Agencia. La evaluación
positiva de un medicamento supondrá la expedición de un certificado
de cumplimiento de la legislación comunitaria relativo al archivo principal
sobre plasma, que irá acompañado del informe de evaluación.
El certificado que se expedirá será válido en toda la Comunidad.
- El archivo principal sobre plasma se actualizará y certificará
de nuevo anualmente.
- Las modificaciones introducidas posteriormente en la formulación del
archivo principal sobre plasma deberán seguir el procedimiento de evaluación
establecido en el Reglamento (CE) n° 542/95(12), relativo al examen de las
modificaciones de los términos de las autorizaciones de comercialización
pertenecientes al ámbito de aplicación del Reglamento (CEE) n°
2309/93 del Consejo de 22 de julio de 1993, por el que se establecen procedimientos
comunitarios para la autorización y supervisión de medicamentos
de uso humano y veterinario y por el que se crea la Agencia Europea para la
Evaluación de Medicamentos(13). Las condiciones para la evaluación
de dichas modificaciones se establecen en el Reglamento (CE) n° 1085/2003.
- En un segundo paso, tras lo dispuesto en los guiones primero, segundo, tercero
y cuarto, la autoridad competente que conceda o que haya concedido la autorización
de comercialización tendrá en cuenta la certificación,
la recertificación o las modificaciones del archivo principal sobre plasma
sobre los medicamentos de que se trate.
- No obstante lo dispuesto en el segundo guión del presente punto (evaluación
y certificación), en caso de que un archivo principal sobre plasma corresponda
únicamente a medicamentos derivados de sangre o plasma cuya autorización
de comercialización se limite a un solo Estado miembro, la evaluación
científica y técnica de dicho archivo principal sobre plasma deberá
realizarla la autoridad nacional competente de ese Estado miembro.
1.2. Vacunas
Respecto a las vacunas de uso humano y no obstante lo dispuesto en el módulo
3 sobre "principio(s) activo(s)", serán de aplicación
los siguientes requisitos, cuando se basen en la utilización de un sistema
de archivo principal sobre antígenos de la vacuna.
El expediente de solicitud de autorización de comercialización
de toda vacuna distinta de la de la gripe humana deberá incluir un archivo
principal sobre cada antígeno de la vacuna que constituya un principio
activo de la misma.
a) Principios
A efectos del presente anexo:
- Se entenderá por "archivo principal sobre un antígeno de
vacuna" una parte independiente del expediente de solicitud de autorización
de comercialización de una vacuna que contendrá toda la información
pertinente de naturaleza biológica, farmacéutica y química
relativa a cada una de los principios activos que forman parte del medicamento.
La parte independiente podrá ser común a una o varias vacunas
monovalentes y/o combinadas que presente el mismo solicitante o titular de autorización
de comercialización.
- Cada vacuna puede contener uno o varios antígenos distintos. Cada vacuna
contiene tantos principios activos como antígenos.
- Una vacuna combinada contiene como mínimo dos antígenos distintos
destinados a la prevención de una o varias enfermedades infecciosas.
- Una vacuna monovalente contiene un solo antígeno destinado a la prevención
de una sola enfermedad infecciosa.
b) Contenido
El archivo principal sobre antígeno de vacuna contendrá la información
siguiente extraída de la parte correspondiente (sustancia activa) del
módulo 3 sobre "calidad de los datos", tal como se define en
la parte I del presente anexo:
Principio activo
1. Información general, incluido el seguimiento de las monografías
pertinentes de la Farmacopea europea.
2. Información sobre la fabricación del principio activo: ha de
abarcar el proceso de fabricación, la información sobre los materiales
de partida y las materias primas, las medidas específicas sobre evaluación
de la seguridad respecto a las EET y los agentes extraños/ externose
instalaciones y equipo.
3. Caracterización del principio activo
4. Control de calidad del principio activo
5. Estándares y materiales de referencia
6. Envase y sistema de cierre del principio activo
7. Estabilidad del principio activo.
c) Evaluación y certificación
- En el caso de las nuevas vacunas que contengan un nuevo antígeno, el
solicitante presentará a una autoridad competente un expediente completo
de solicitud de autorización de comercialización que incluya todos
los archivos principales sobre antígeno de vacuna correspondientes a
cada uno de los antígenos que forman parte de la nueva vacuna, en el
caso de que no exista ya ningún archivo principal de cada antígeno.
La Agencia realizará la evaluación científica y técnica
del mencionado archivo principal sobre antígeno de vacuna. La evaluación
positiva de un medicamento supondrá la expedición de un certificado
de cumplimiento de la legislación comunitaria relativo a cada archivo
principal sobre antígeno de vacuna, que irá acompañado
del informe de evaluación. El certificado que se expedirá tendrá
validez en toda la Comunidad.
- Lo dispuesto en el primer guión será aplicable a cada vacuna
consistente en una nueva combinación de antígenos de vacuna, independientemente
de que alguno de dichos antígenos pueda formar parte de vacunas ya autorizadas
en la Comunidad.
- Las modificaciones del contenido de un archivo principal sobre antígeno
de vacuna correspondiente a una vacuna autorizada en la Comunidad estarán
sujetas a una evaluación científica y técnica que realizará
la Agencia con arreglo al procedimiento establecido en el Reglamento (CE) n°
1085/2003. En caso de evaluación positiva, la Agencia expedirá
un certificado de cumplimiento de la legislación comunitaria del archivo
principal sobre el antígeno de vacuna. El certificado que se expedirá
tendrá validez en toda la Comunidad.
- No obstante lo dispuesto en el primero, segundo y tercer guiones del presente
apartado (evaluación y certificación), en caso de que un archivo
principal sobre antígeno de vacuna corresponda únicamente a una
vacuna que ha sido objeto de una autorización de comercialización
que no se ha concedido (o que no se concederá) según un procedimiento
comunitario y siempre que la vacuna autorizada incluya antígenos de vacuna
no evaluados mediante un procedimiento comunitario, la autoridad nacional competente
que concedió la autorización de comercialización realizará
la evaluación científica y técnica del mencionado archivo
principal sobre antígeno de vacuna y sus posteriores modificaciones.
- En un segundo paso, tras lo dispuesto en los guiones primero, segundo, tercero
y cuarto, la autoridad competente que conceda o que haya concedido la autorización
de comercialización tendrá en cuenta la certificación,
la recertificación o las modificaciones del archivo principal sobre antígeno
de vacuna relativa a los medicamentos de que se trate.
2. RADIOFÁRMACOS Y PRECURSORES
2.1. Radiofármacos
A efectos del presente capítulo, para las solicitudes basadas en el
apartado 2 del artículo 6 y el artículo 9 deberá presentarse
un expediente completo en el que se incluirán los siguientes datos específicos:
Módulo 3
a) Cuando se trate de dispositivos reactivos radiofarmaceúticos que
deban ser marcados radiactivamente tras el suministro por el fabricante, se
considerará que el principio activo es aquella parte de la formulación
cuyo propósito es transportar o unirse al radionucleido. La descripción
del método de fabricación de equipos reactivos radiofarmaceúticos
incluirá detalles de la fabricación del equipo y del tratamiento
final recomendado para producir el radiofármaco. Las especificaciones
necesarias del radionucleido se describirán, si es pertinente, con arreglo
a la monografía general o las monografías específicas de
la Farmacopea Europea. Además, se describirá cualquier compuesto
esencial para el marcado radiactivo. También se describirá la
estructura del compuesto al que se ha aplicado el marcado radiactivo.
En cuanto a los radionucleido, se discutirán las reacciones nucleares
que comportan.
En un generador, tanto los radionucleido padre como hijo ( en inglés
se indica madre e hija) se considerarán sustancias activas.
b) Se ofrecerán detalles sobre la naturaleza del radionúclido,
la identidad del isótopo, las impurezas probables, el portador, el uso
y la actividad específica.
c) Las materias objeto de irradiación se incluyen entre los materiales
de partida.
d) Se incluirán consideraciones acerca de la pureza química/radioquímica
y su relación con la biodistribución.
e) Se describirá la pureza radionucleídica, la pureza radioquímica
y la actividad específica.
f) Para los generadores se requiere información detallada sobre las pruebas
de radionucleido padre e hijo (en inglés se indica madre e hija). En
el caso de eluidos del generador deben realizarse pruebas de radionucleido padres
(en inglés madre) y de los demás componentes del generador.
g) El requisito de expresar el contenido en sustancias activas en términos
de masa de las fracciones activas sólo se aplicará a los equipos
reactivos (kits) radiofarmacéuticos. Cuando se trate de radionucleido
la radiactividad se expresará en bequerelios, fijando la fecha y, si
fuera necesario, la hora, haciendo referencia al huso horario. Deberá
indicarse el tipo de radiación.
h) En el caso de dispositivos reactivos, las especificaciones del producto acabado
incluirán pruebas del funcionamiento de los productos tras el marcado
radiactivo. Deberán incluirse controles apropiados de pureza radioquímica
y radionucleídica del producto marcado radiactivamente. Se identificarán
y controlaran todos los materiales esenciales para el marcado radiactivo.
i) Se ofrecerá información sobre la estabilidad en el caso de
los generadores radionucleídicos, los dispositivos radionucleídicos
y los productos marcados radiactivamente. La estabilidad durante el uso de los
radiofármacos en viales multidosis, deberá documentarse.
Módulo 4
Se estima que la toxicidad puede ir asociada a la dosis de radiación.
En diagnóstico, esto es consecuencia del uso de radiofármacos;
en terapia, es la propiedad deseada. Por tanto, la evaluación de la seguridad
y eficacia de los radiofármacos tendrá en cuenta los requisitos
de los medicamentos y los aspectos de la dosimetría de la radiación.
Deberá documentarse la exposición a las radiaciones de órganos
y tejidos. Las estimaciones de las dosis de radiación absorbida se calcularán
con arreglo a un sistema especificado, reconocido internacionalmente mediante
una determinada vía de administración.
Módulo 5
Cuando proceda, se facilitarán los resultados de los ensayos clínicos;
si no se hace, deberá justificarse en las síntesis clínicas.
2.2. Precursores radiofarmacéuticos con fines de marcado radiactivo
En el caso específico de un precursor radiofarmacéutico que tenga
únicamente por objeto el marcado radiactivo, el objetivo primario será
presentar información acerca de las posibles consecuencias de una escasa
eficiencia del marcado radiactivo o de la disociación in vivo de la entidad
conjugada marcada radiactivamente, es decir, las cuestiones relacionadas con
las repercusiones en el paciente del radionucleido libre. Por otra parte, también
es necesario presentar toda la información pertinente en relación
con los riesgos profesionales, como la exposición a la radiación
para el personal hospitalario y el entorno.
En concreto, se facilitará cuando proceda la información que se
especifica a continuación:
Módulo 3
Lo dispuesto en el módulo 3, definido en las letras a) a i), se aplicará,
cuando proceda, al registro de los precursores radiofarmacéuticos.
Módulo 4
En lo que respecta a la toxicidad por dosis única y por administración
continuada, deberán facilitarse (a menos que se justifique el motivo
por el que no se hace) los resultados de los estudios realizados de conformidad
con los principios de las buenas prácticas de laboratorio que establecen
las Directivas 87/18/CEE y 88/320/CEE del Consejo.
Los estudios de mutagenicidad sobre el radionúclido no se consideran
útiles en este caso concreto.
Deberá presentarse información relacionada con la toxicidad química
y la disposición del núclido "frío" pertinente.
Módulo 5
La información clínica elaborada a partir de estudios clínicos
mediante el precursor en sí no se considera pertinente en el caso específico
de un precursor radiofarmacéutico que tenga únicamente por objeto
el marcado radiactivo.
No obstante, se facilitará información que demuestre la utilidad
clínica del precursor radiofarmacéutico cuando está ligado
a moléculas portadoras pertinentes.
3. MEDICAMENTOS HOMEOPÁTICOS
En este apartado se exponen las disposiciones específicas sobre la aplicación
de los módulos 3 y 4 a los medicamentos homeopáticos definidos
en el apartado 5 del artículo 1.
Módulo 3
Lo dispuesto en el módulo 3 se aplicará a los documentos presentados
conforme al artículo 15 en el registro simplificado de medicamentos homeopáticos
que se mencionan en el apartado 1 del artículo 14, así como a
los documentos para la autorización de otros medicamentos homeopáticos
mencionados en el apartado 1 del artículo 16, con las modificaciones
que se exponen a continuación.
a) Terminología
La denominación latina de la cepa homeopática descrita en el expediente
de solicitud de autorización de comercialización deberá
ser acorde con la denominación latina de la Farmacopea Europea o, en
su ausencia, con la de una farmacopea oficial de un Estado Miembro. Se incluirá,
cuando resulte pertinente, el nombre o nombres tradicionales usados en cada
Estado Miembro.
b) Control de los materiales de partida
Los detalles y documentos sobre los materiales de partida, es decir, todos los
materiales utilizados incluidas las materias primas e intermedias hasta la dilución
final que haya de incorporarse al producto terminado, que se adjunten a la solicitud,
se complementarán con datos adicionales sobre la cepa homeopática.
Los requisitos generales de calidad se aplicarán a todos los materiales
de partida y materias primas, así como a los pasos intermedios del proceso
de fabricación hasta la dilución final que será incorporada
al producto terminado. Si es posible, se requerirá una determinación
cuantitativa si hay presencia de componentes tóxicos y si, debido al
elevado grado de dilución, la calidad no puede ser controlada en la dilución
final que será incorporada. Se describirá minuciosamente cada
paso del proceso de fabricación desde los materiales de partida hasta
la dilución final que será incorporada al producto terminado.
En el caso de intervención de diluciones, dichos pasos de dilución
deberán realizarse de acuerdo con los métodos homeopáticos
de fabricación establecidos en la monografía pertinente de la
Farmacopea Europea o, en su defecto, en una farmacopea oficial de un Estado
Miembro.
c) Métodos de control del producto terminado
Los requisitos generales de calidad serán aplicables a los medicamentos
homeopáticos acabados; el solicitante deberá justificar debidamente
cualquier excepción.
Se efectuará la identificación y determinacion cuantitativa de
todos los componentes pertinentes desde un punto de vista toxicológico.
En caso de que pueda justificarse que no es posible la identificación
y/o la cuantificación de todos los componentes pertinentes desde un punto
de vista toxicológico (por ejemplo, debido a su dilución en el
producto terminado), la calidad deberá demostrarse mediante la validación
completa de los procesos de fabricación y dilución.
d) Pruebas de estabilidad
Deberá demostrarse la estabilidad del producto terminado. Los datos de
estabilidad de las cepas homeopáticas generalmente son transferibles
a las diluciones/trituraciones obtenidas de las mismas. Si no es posible la
identificación o determinación cuantitativa del principio activo
debido al grado de dilución, podrán considerarse los datos de
estabilidad de la forma farmacéutica.
Módulo 4
Las disposiciones del presente módulo serán aplicables al registro
simplificado de los medicamentos homeopáticos mencionados en el apartado
1 del artículo 14, con las especificaciones siguientes.
Se justificará la ausencia de cualquier dato; por ejemplo, se justificará
por qué puede afirmarse la existencia de un nivel aceptable de seguridad,
pese a la ausencia de determinados estudios.
4. MEDICAMENTOS A BASE DE PLANTAS
Las solicitudes relativas a medicamentos a base de plantas se presentarán
con un expediente completo en el que figurarán los detalles específicos
siguientes.
Módulo 3
Lo dispuesto en el módulo 3, incluido el seguimiento de las monografías
pertinentes de la Farmacopea Europea, se aplicará a la autorización
de medicamentos a base de plantas. Al presentar la solicitud se tendrá
en cuenta el estado de los conocimientos científicos.
Habrán de considerarse los siguientes aspectos específicos de
los medicamentos a base de plantas.
(1) Sustancias y preparados vegetales
A efectos del presente anexo, el término "sustancias vegetales y
preparados vegetales" se considerará equivalente al término
"herbal drugs and herbal drug preparations", tal y como aparece definido
en la Farmacopea Europea.
Respecto a la nomenclatura de las sustancias vegetales, se incluirá la
denominación científica binomial de la planta (género,
especie, variedad y autor), así como su quimiotipo (cuando proceda),
las partes de las plantas utilizadas, la definición de la sustancia vegetal,
los otros nombres (sinónimos mencionados en las otras farmacopeas) y
el código de laboratorio.
Respecto a la nomenclatura del preparado vegetal, se incluirá la denominación
científica binomial de la planta (género, especie, variedad y
autor), así como su quimiotipo (cuando proceda), las partes de las plantas
utilizadas, la definición del preparado vegetal, la proporción
entre la sustancia vegetal y el preparado vegetal, el (los) disolvente(s) para
extracción, otros nombres (sinónimos mencionados en otras farmacopeas)
y el código de laboratorio.
Para documentar el apartado de la estructura de la(s) sustancia(s) vegetal(es)
y el (los) preparado(s) vegetal(es) cuando proceda, se incluirán la forma
física, la descripción de los componentes con actividad terapéutica
conocida o los marcadores (fórmula molecular, masa molecular relativa,
fórmula estructural, incluidas la estereoquímica relativa y absoluta,
la fórmula molecular y la masa molecular relativa), así como las
de otros constituyentes.
Con el fin de documentar el apartado sobre el fabricante de la sustancia vegetal,
se incluirán, cuando proceda, el nombre, la dirección y la responsabilidad
de cada proveedor, incluidos contratistas, y cada lugar o instalación
propuestos para la producción/recogida y control de la sustancia vegetal.
Con el fin de documentar el apartado sobre el fabricante del preparado vegetal,
se incluirán, cuando proceda, el nombre, la dirección y la responsabilidad
de cada fabricante, incluidos contratistas, y cada lugar de fabricación
o instalación propuestos para la fabricación y ensayo del preparado
vegetal.
En relación con la descripción del proceso de fabricación
y de los controles del proceso de la sustancia vegetal, se ofrecerá información
para describir adecuadamente la producción y recogida de plantas, incluidas
la procedencia geográfica de la planta medicinal y sus condiciones de
cultivo, cosecha, secado y almacenamiento.
En relación con la descripción del proceso de fabricación
y de los controles del proceso del preparado vegetal, se ofrecerá información
para describir adecuadamente el proceso de fabricación del preparado
vegetal, incluida la descripción del tratamiento, los disolventes y reactivos,
las fases de purificación y la estandarización.
Por lo que se refiere al desarrollo del proceso de fabricación, se presentará
cuando proceda un breve resumen en el que se describa el desarrollo de la(s)
sustancia(s) vegetal(es) y el (los) preparado(s) vegetal, teniendo en cuenta
la vía de administración y utilización propuestas. Deberán
discutirse, cuando proceda, los resultados en que se compare la composición
fitoquímica de las sustancias vegetales y preparados vegetal(es), según
el caso, reseñado(s) en los datos bibliográficos de apoyo y las
sustancias vegetales y preparados vegetales, según el caso, que contiene
como sustancias activas el medicamento a base de plantas para el que se solicita
la autorización.
Respecto a la dilucidación de la estructura y otras características
de la sustancia vegetal(es), se facilitará información sobre la
caracterización botánica, macroscópica, microscópica
y fitoquímica, así como sobre su actividad biológica si
fuera necesario.
Respecto a la dilucidación de la estructura y otras características
de los preparados vegetales, se facilitará información sobre la
caracterización fitoquímica y físico-química, así
como sobre su actividad biológica si fuera necesario.
Se presentarán cuando proceda las especificaciones de la(s) sustancia(s)
vegetal(es)y de los preparado(s) vegetales.
También se informará si procede acerca de los procedimientos analíticos
empleados para controlar la(s) sustancia(s) vegetal(es)y los preparado(s) vegetal(es).
Por lo que se refiere a la validación de los procedimientos analíticos,
cuando proceda, se ofrecerá información sobre validación
analítica, incluyendo los datos experimentales de los procedimientos
analíticos empleados para controlar la(s) sustancia(s) vegetal(es)y los
preparado(s) vegetal(es).
En relación con el análisis de los lotes, se describirán
si procede los lotes y los resultados de los análisis de los mismos en
relación con la(s) sustancia(s) vegetal(es)y los preparado(s) vegetal(es),
incluyendo los de las sustancias de farmacopea.
Habrán de justificarse, cuando sea pertinente, las especificaciones de
la(s) sustancia(s) vegetal(es) y de los preparado(s) vegetal(es).
Asimismo se informará, en su caso, sobre las normas y materiales de referencia
empleados para probar la(s) sustancia(s) vegetal(es) y los preparado(s) vegetal(es).
Cuando la sustancia vegetal o el preparado vegetal sea objeto de una monografía,
el solicitante podrá solicitar un certificado de idoneidad expedido por
la Dirección Europea de la Calidad del Medicamento.
(2) Medicamentos a base de plantas
Respecto al desarrollo de la formulación, se presentará un resumen
sucinto en el que se describirá el desarrollo del medicamento a base
de plantas, teniendo en cuenta la vía de administración y la utilización
propuestas. Deberán discutirse, cuando proceda, los resultados en los
que se compare la composición fitoquímica de los productos reseñados
en los datos bibliográficos de apoyo y el medicamento a base de plantas
para el que se solicita autorización.
5. MEDICAMENTOS HUÉRFANOS
- En el caso de un medicamento huérfano en el sentido del Reglamento
(CE) n° 141/2000, se pueden aplicar las disposiciones generales que figuran
en el punto 6 de la parte II (circunstancias excepcionales). El solicitante
deberá justificar en los resúmenes no clínicos y clínicos
las razones que impiden facilitar la información completa, así
como el balance riesgo/beneficios del medicamento huérfano de que se
trate.
- Cuando un solicitante de una autorización de comercialización
de un medicamento huérfano invoque lo dispuesto en el inciso ii) de la
letra a) del apartado 1 del artículo 10 y en el punto 1 de la parte II
del presente anexo (uso médico suficientemente comprobado), la utilización
sistemática y documentada de la sustancia de que se trate puede referirse,
con carácter excepcional, a la utilización de dicha sustancia
conforme a lo dispuesto en el artículo 5 de la presente Directiva.
PARTE IV
MEDICAMENTOS DE TERAPIA AVANZADA
Los medicamentos de terapia avanzada se basan en procesos de fabricación
que se basan en diversas moléculas biológicas producidas por transferencia
genética y/o en células terapéuticas modificadas biológicamente
avanzados como sustancias activas o parte de las mismas.
La presentación de la solicitud de autorización de comercialización
de dichos medicamentos deberá satisfacer los requisitos de formato expuestos
en la parte I del presente anexo.
Los módulos 1 a 5 serán de aplicación. En cuanto a los
organismos modificados genéticamente liberados intencionalmente en el
medio ambiente, deberá prestarse atención a la persistencia de
los OMG en el destinatario y la posible duplicación y/o modificación
de los mismos al liberarse en el medio ambiente. La información relativa
al riesgo para el medio ambiente deberá figurar en el anexo del módulo
1.
1. MEDICAMENTOS DE TERAPIA GÉNICA (DE ORIGEN HUMANO Y XENOGÉNICOS)
A efectos del presente anexo, se entenderá por medicamento de terapia
génica un producto obtenido mediante un conjunto de procesos de fabricación
destinados a transferir, bien in vivo bien ex vivo, un gen profiláctico,
de diagnóstico o terapéutico (es decir, un trozo de ácido
nucleico) a células humanas/animales y su posterior expresión
in vivo. La transferencia genética supone un sistema de expresión
contenido en un sistema de distribución conocido como vector, que puede
ser de origen viral o no viral. El vector puede incluirse asimismo en una célula
humana o animal.
1.1. Diversidad de los medicamentos de terapia génica
a) Medicamentos de terapia génica basados en células alogénicas
o xenogénicas
El vector se prepara para su utilización y se almacena antes de transferirlo
a las células huéspedes.
Las células se han obtenido anteriormente y pueden tratarse como un banco
de células (recogida en banco o banco establecido mediante la obtención
de células primarias) con una viabilidad limitada.
Las células modificadas genéticamente por el vector representan
un principio activo.
Pueden darse otros pasos suplementarios para obtener el producto terminado.
Esencialmente, el objeto de un medicamento de este tipo es su administración
a una determinada cantidad de pacientes.
b) Medicamentos de terapia génica en los que se utilizan células
humanas autólogas
El principio activo es un lote de vector preparado para su utilización
que se almacena antes de transferirlo a las células autólogas.
Pueden darse otros pasos suplementarios para obtener el producto terminado.
Estos productos se preparan a partir de células obtenidas de un solo
paciente. A continuación las células se modifican genéticamente
mediante un vector preparado para su utilización que contiene el gen
adecuado que se ha preparado con antelación y constituye el principio
activo. La preparación, destinada por definición a un solo paciente,
se le reinyecta al paciente. La totalidad del proceso de fabricación,
desde la recogida de las células del paciente hasta la reinyección
al paciente, se considerará una sola intervención.
c) Administración de vectores preparados para su utilización
con material genético insertado (profilático, de diagnóstico
o terapéutico)
El principio activo es un lote de vector preparado para su utilización.
Pueden darse otros pasos suplementarios para obtener el producto terminado.
Este tipo de medicamento se destina a su administración a varios pacientes.
La transferencia de material genético puede realizarse por inyección
directa del vector preparado para su utilización a sus destinatarios.
1.2. Requisitos específicos relativos al módulo 3
Entre los medicamentos de terapia génica se encuentran los siguientes:
- ácido nucleico desnudo
- ácido nucleico complejado o vectores no virales
- vectores virales
- células modificadas genéticamente.
En cuanto a los demás medicamentos, pueden establecerse los tres elementos
principales del proceso de fabricación, a saber:
- los materiales de partida: los materiales a partir de los que se fabrica el
principio activo, como el gen de que se trate, los plásmidos de expresión,
los bancos de células y los stocks de virus o vector no viral;
- principio activo: vector recombinante, virus, plásmidos desnudos o
complejos, virus productores de células, células modificadas genéticamente
in vitro;
- Producto terminado: principio activo formulado en su recipiente primario final
para el uso médico previsto. En función del tipo de medicamento
de terapia génica, la vía de administración y las condiciones
de utilización pueden requerir el tratamiento ex vivo de las células
del paciente (véase 1.1.b).
Deberá prestarse particular atención a los elementos siguientes.
a) Se facilitará información sobre las características
pertinentes del medicamento de terapia génica, incluida su expresión
en la población celular destinataria. Se facilitará información
sobre la fuente, construcción, caracterización y verificación
de la secuencia genética codificante, incluidas su integridad y estabilidad.
Se facilitará la secuencia completa de otros genes, los elementos reguladores
y el esqueleto del vector.
b) Se ofrecerá información acerca de la caracterización
del vector utilizado para transferir y transportar el gen. Ello incluirá
su caracterización físico-química y/o biológica/inmunológica.
Para los medicamentos que utilicen un microorganismo, como las bacterias o los
virus, para facilitar la transferencia genética (transferencia genética
biológica), datos sobre la patogénesis de la cepa parental y sobre
su tropismo para tipos específicos de tejidos y células, así
como la dependencia de la interacción respecto al ciclo celular.
Por lo que respecta a los medicamentos que empleen medios no biológicos
para facilitar la transferencia génica, deberán comunicarse las
propiedades físico-químicas de los componentes individualmente
y combinados.
c) Los principios de establecimiento y caracterización de bancos de células
o lotes de siembra se aplicarán, en su caso, a los medicamentos producidos
mediante transferencia génica.
d) Deberá comunicarse la fuente de las células que albergan el
vector recombinante.
Se documentarán las características de la fuente humana, como
la edad, el sexo, los resultados de las pruebas microbiológicas y virales,
los criterios de exclusión y el país de origen.
Para las células de origen animal, se ofrecerán datos detallados
relativos a los elementos siguientes:
- Origen de los animales
- Cría y cuidado de los animales
- Animales transgénicos (métodos de creación, caracterización
de las células transgénicas, naturaleza del gen insertado)
- Medidas para prevenir y controlar las infecciones en los animales fuente/donantes
- Pruebas relativas a agentes infecciosos
- Instalaciones
- Control de los materiales de partida y las materias primas.
Deberá documentarse la metodología de la recogida de células,
incluyendo el lugar, el tipo de tejido, el proceso operativo, el agrupamiento,
el almacenamiento y la trazabilidad, así como los controles realizados
durante la recogida.
e) La evaluación de la seguridad viral y la trazabilidad de los productos
desde el donante al producto terminado constituyen partes fundamentales de la
documentación que se ha de presentar. Por ejemplo, se excluirá
la presencia de virus competentes para replicación en stocks de vectores
virales no replicativos.
2. MEDICAMENTOS DE TERAPIA CELULAR SOMÁTICA (DE ORIGEN HUMANO Y XENOGÉNICOS)
A efectos del presente anexo, se entenderá por medicamentos de terapia
celular somática la utilización en seres humanos de células
somáticas vivas, tanto autólogas (procedentes del propio paciente),
como alogénicas (de otro ser humano) o xenogénicas (de animales),
cuyas características biológicas han sido alteradas sustancialmente
como resultado de su manipulación para obtener un efecto terapéutico,
de diagnóstico o preventivo por medios metabólicos, farmacológicos
e inmunológicos. Dicha manipulación incluye la expansión
o activación de poblaciones celulares autólogas ex vivo (p. ej.,
inmunoterapia adoptiva), la utilización de células alogénicas
y xenogénicas asociadas con productos sanitarios empleados ex vivo o
in vivo (p. ej., microcápsulas, matrices y andamiajes intrínsecos,
biodegradables o no biodegradables).
Requisitos especiales para medicamentos de terapia celular en relación
con el módulo 3
Entre los medicamentos de terapia celular somática se encuentran los
siguientes:
- Células manipuladas para modificar sus propiedades inmunológicas,
metabólicas o funcionales de otro tipo en aspectos cualitativos o cuantitativos.
- Células clasificadas, seleccionadas y manipuladas, que se someten posteriormente
a un proceso de fabricación con el fin de obtener el producto terminado.
- Células manipuladas y combinadas con componentes no celulares (por
ejemplo, matrices o productos sanitarios biológicos o inertes) que ejercen
la acción pretendida en principio en el producto acabado.
- Derivados de células autólogas expresadas in vitro en condiciones
específicas de cultivo.
- Células modificadas genéticamente o sometidas a otro tipo de
manipulación para expresar propiedades funcionales homólogas o
no homólogas anteriormente no expresadas.
La totalidad del proceso de fabricación, desde la recogida de las células
del paciente (situación autóloga) hasta la reinyección
al paciente, se considerará una sola intervención.
En cuanto a los demás medicamentos, pueden establecerse los tres elementos
del proceso de fabricación, a saber:
- los materiales de partida: los materiales a partir de los que se fabrica la
sustancia activa, esto es, órganos, tejidos, fluidos corporales o células;
- los principios activos: células manipuladas, lisados celulares, células
proliferantes y células utilizadas junto con matrices y productos sanitarios
inertes;
- los productos terminados: principio activo formulado en su recipiente primario
final para el uso médico previsto.
a) Información general sobre el (los) principio(s) activo(s)
Los principios activos de los medicamentos de terapia celular consisten en células
que, como consecuencia del tratamiento in vitro, presentan propiedades profilácticas,
de diagnóstico o terapéuticas distintas de sus propiedades fisiológicas
y biológicas originales.
En este apartado se describirá el tipo de células y cultivo correspondientes.
Se documentarán los tejidos, órganos o fluidos biológicos
de los que derivan las células, así como la naturaleza autóloga,
alogénica o xenogénica de la donación y su origen geográfico.
Se detallará la recogida de células, el muestreo y el almacenamiento
previo a transformaciones posteriores. En el caso de las células alogénicas,
se prestará una atención especial a la primera fase del proceso,
que incluye la selección de donantes. Deberá informarse acerca
del tipo de manipulación realizada y la función fisiológica
de las células que se utilizan como sustancia activa.
b) Información relacionada con los materiales de partida del (los)
principio(s) activo(s)
1. Células somáticas humanas
Los medicamentos de terapia con células somáticas humanas están
formados por un número definido (pool) de células viables que
derivan de un proceso de fabricación que comienza bien en el nivel de
los órganos o tejidos recuperados de un ser humano, bien en el nivel
de un sistema de banco de células muy definido, en el que el pool de
células se basa en líneas continuas de células. A efectos
del presente capítulo, se entenderá por principio activo el pool
de semillas de células humanas y por producto terminado el pool de semillas
de células humanas formuladas para el uso médico previsto.
Se documentarán minuciosamente los materiales de partida y cada etapa
del proceso de fabricación, incluyendo los aspectos de seguridad viral.
(1) Órganos, tejidos, células y fluidos corporales de origen
humano
Se documentarán las características de la fuente humana, como
la edad, el sexo, la situación microbiológica, los criterios de
exclusión y el país de origen.
Deberá documentarse la descripción del muestreo, incluyendo el
lugar, el tipo, el proceso operativo, el agrupamiento, el almacenamiento y la
trazabilidad, así como los controles realizados.
(2) Sistemas de bancos celulares
Los requisitos pertinentes descritos en la parte I serán de aplicación
para la preparación y el control de calidad de los sistemas de bancos
de células. Ello se refiere básicamente a las células alogénicas
o xenogénicas.
(3) Materiales o productos sanitarios auxiliares
Se proporcionará información sobre la utilización de cualquier
materia prima (p. ej., citoquinas, factores de crecimiento, medios de cultivo)
o de posibles productos sanitarios auxiliares, (p. ej, productos de clasificación
de células, matriz de polímeros biocompatible, fibras, cuentas),
por lo que se refiere a su biocompatibilidad, funcionalidad y al riesgo de agentes
infecciosos.
2. Células somáticas animales (xenogénicas)
Se facilitará información pormenorizada en relación con
los siguientes elementos:
- Origen de los animales
- Cría y cuidado de los animales
- Animales modificados genéticamente (métodos de creación,
caracterización de las células transgénicas, naturaleza
del gen insertado o eliminado)
- Medidas para prevenir y controlar las infecciones en los animales fuente/donantes
- Pruebas relativas a agentes infecciosos, incluidos microorganismos transmitidos
verticalmente (también retrovirus endógenos)
- Instalaciones
- Sistemas de bancos celulares
- Control de los materiales de partida y las materias primas.
a) Información sobre el proceso de fabricación del principio(s)
activo(s) y el producto acabado
Deberán documentarse los distintos pasos del proceso de fabricación,
tales como la disociación órgano/tejido, la selección de
la población celular que interese, el cultivo de la célula in
vitro, y la transformación de la misma bien mediante agentes físico-químicos,
bien por transferencia génica.
b) Caracterización del principio(s) activo(s)
Se proporcionará toda la información pertinente sobre la caracterización
de la población celular de que se trate desde el punto de vista de la
identidad (especie de origen, citogenética de bandeo, análisis
morfológico), pureza (agentes microbianos extraños/externos y
contaminantes celulares), potencia (actividad biológica definida) y adecuación
(pruebas de cariología y tumorigenicidad) para el uso médico previsto.
c) Desarrollo farmacéutico del producto terminado
Además del método de administración específico utilizado
(infusión intravenosa, inyección en el lugar de la lesión,
cirugía de trasplantes), deberá ofrecerse información sobre
la utilización de posibles productos sanitarios auxiliares (polímeros
biocompatibles, matriz, fibras, cuentas), por lo que se refiere a su compatibilidad
y durabilidad.
d) Trazabilidad
Se presentará un diagrama detallado para asegurar la trazabilidad de
los productos desde el donante hasta el producto terminado.
3. REQUISITOS ESPECÍFICOS DE LOS MEDICAMENTOS DE TERAPIA GÉNICA
Y TERAPIA CELULAR SOMÁTICA (DE ORIGEN HUMANO Y XENOGÉNICOS) EN
RELACIÓN CON LOS MÓDULOS 4 Y 5
3.1. Módulo 4
Para los medicamentos de terapia génica y somática, se reconoce
que los requisitos convencionales establecidos en el módulo 4 para las
pruebas no clínicas de medicamentos no siempre resultan adecuados, debido
a las propiedades estructurales y biológicas, únicas y diversas,
de tales productos, que abarcan un elevado grado de especificidad según
la especie y el sujeto, barreras inmunológicas y diferencias en las respuestas
pleiotrópicas.
Deberán ilustrarse adecuadamente en el módulo 2 las razones que
aconsejan el desarrollo no clínico y los criterios aplicados para elegir
las especies y modelos pertinentes.
Puede resultar necesario identificar o desarrollar nuevos modelos animales con
el fin de contribuir a la extrapolación de conclusiones específicas
sobre parámetros funcionales y toxicidad para la actividad in vivo de
los productos en los seres humanos. Deberá justificarse científicamente
la utilización de dichos modelos animales de dolencia para apoyar la
seguridad y la prueba del concepto en aras de la eficacia.
3.2. Módulo 5
Deberá demostrarse y describirse en el módulo 5 la eficacia de
los medicamentos de terapia avanzada. No obstante, en el caso de determinados
productos e indicaciones terapéuticas, quizá no resulte posible
realizar ensayos clínicos convencionales. En el módulo 2 se justificará
cualquier desviación respecto a las directrices vigentes.
El desarrollo clínico de los medicamentos de terapia avanzada tendrán
algunos rasgos especiales debido a la complejidad y labilidad de los principios
activos. Requiere consideraciones suplementarias, a causa de las cuestiones
relacionadas con la viabilidad, proliferación, migración y diferenciación
de las células (terapia celular somática) y su capacidad de crecer
y diferenciarse (terapia celular), y de las especiales circunstancias clínicas
en las que se utilizan los productos o del especial modo de acción mediante
la expresión genética (terapia génica somática).
En la solicitud de autorización de medicamentos de terapia avanzada deberán
abordarse los riesgos especiales relacionados con dichos productos, derivados
de la contaminación potencial con agentes infecciosos. Deberá
hacerse especial hincapié tanto en las fases tempranas de desarrollo,
incluida la elección de los donantes en el caso de los medicamentos de
terapia celular, como en la intervención terapéutica en su conjunto,
incluidos un manejo y administración adecuados del producto.
Por otra parte, en el módulo 5 de la solicitud deberán incluirse,
si procede, datos sobre las medidas de inspección y control de las funciones
y el desarrollo de las células vivas en el destinatario, con el fin de
impedir la transmisión de agentes infecciosos a este y minimizar todos
los posibles riesgos para la salud pública.
3.2.1. Estudios de farmacología humana y eficacia
Los estudios de farmacología humana deberán ofrecer información
sobre el modo de acción y la eficacia que se prevén según
parámetros justificados, la biodistribución, la dosificación
adecuada, el calendario y los métodos de administración o modalidad
de utilización deseable para los estudios de eficacia.
Los estudios de farmacocinética convencionales pueden no ser pertinentes
para determinados productos de terapia avanzada. En ocasiones, los estudios
en voluntarios sanos no son viables y el establecimiento de la dosificación
y la cinética resultará difícil de determinar en los ensayos
clínicos. No obstante, es necesario estudiar la distribución y
el comportamiento in vivo del producto, incluyendo la proliferación de
las células y la función a largo plazo, así como el alcance
y distribución del producto génico y la duración de la
expresión génica deseada. Deberá recurrirse a pruebas adecuadas
y, en caso necesario, se desarrollarán para rastrear el producto celular
o la célula que exprese el gen deseado en el cuerpo humano y para controlar
la función de las células que se administraron o transfectaron.
La evaluación de la eficacia y seguridad de un medicamento de terapia
avanzada deberá incluir la minuciosa descripción y evaluación
del procedimiento terapéutico en su conjunto, incluidas las vías
especiales de administración (como la transfección de células
ex vivo, la manipulación in vitro o el empleo de técnicas de intervención)
y la detección de posibles regímenes asociados (incluidos el tratamiento
inmunosupresor, antiviral y citotóxico).
Deberá ponerse a prueba el procedimiento en su integridad en ensayos
clínicos y describirse en la información sobre el producto.
3.2.2. Seguridad
Deberán considerarse las cuestiones de seguridad que plantean la respuesta
inmunitaria a los medicamentos o a las proteínas expresadas, el rechazo
inmunitario, la inmunosupresión y el fallo de los dispositivos de inmunoaislamiento.
Determinados medicamentos de terapia génica avanzada y de terapia celular
somática (por ejemplo, productos de terapia celular xenogénica
y algunos de transferencia genética) pueden contener partículas
y agentes infecciosos aptos para la duplicación. Se podrá efectuar
un seguimiento del paciente en lo referente al desarrollo de posibles infecciones
y sus secuelas patológicas durante las fases previa y/o posterior a la
autorización; dicha vigilancia podrá ampliarse a las personas
en contacto con él, incluido el personal sanitario.
Al usar determinados medicamentos de terapia celular somática y de transferencia
genética no puede eliminarse totalmente el riesgo de contaminación
con agentes potencialmente transmisibles. No obstante, el riesgo puede reducirse
al mínimo mediante las medidas descritas en el módulo 3.
Las medidas incluidas en el proceso de producción habrán de complementarse
con métodos de prueba con acompañamiento, procesos de control
de calidad y métodos adecuados de vigilancia que deben describirse en
el módulo 5.
El empleo de determinados medicamentos de terapia celular somática avanzada
podrá limitarse, de manera temporal o permanente, a establecimientos
que hayan documentado competencias especializadas e instalaciones para garantizar
un seguimiento específico de la seguridad de los pacientes. Podrá
resultar pertinente el mismo planteamiento para determinados medicamentos de
terapia génica que se asocian con un riesgo potencial de contener agentes
infecciosos aptos para la duplicación.
En la solicitud también se considerarán y abordarán los
aspectos del seguimiento a largo plazo en relación con complicaciones
tardías.
Cuando proceda, el solicitante deberá presentar el plan detallado de
gestión de riesgos que abarque los datos clínicos y de laboratorio
del paciente, los datos epidemiológicos que surjan y, en su caso, datos
de los archivos de muestras de tejidos del donante y el destinatario. Un sistema
de este tipo es necesario para asegurar la trazabilidad del medicamento y la
rápida respuesta a modelos sospechosos de acontecimientos adversos.
4. DECLARACIÓN ESPECÍFICA SOBRE MEDICAMENTOS DE XENOTRASPLANTE
A efectos del presente anexo, se entenderá por xenotrasplante todo procedimiento
que implique el trasplante, implantación o infusión en un destinatario
humano de tejidos u órganos vivos procedentes de animales, o bien fluidos,
células, tejidos u órganos que hayan experimentado contacto ex
vivo con células, tejidos u órganos vivos animales.
Habrá de prestarse una atención muy especial a los materiales
de partida.
A este respecto, se facilitará información pormenorizada en relación
con los siguientes elementos con arreglo a directrices específicas:
- Origen de los animales
- Cría y cuidado de los animales
- Animales modificados genéticamente [métodos de creación,
caracterización de las células transgénicas, naturaleza
del gen insertado o eliminado (knocked out)]
- Medidas para prevenir y controlar las infecciones en los animales fuente/donantes
- Pruebas relativas a agentes infecciosos
- Instalaciones
- Control de los materiales de partida y materias primas
- Trazabilidad.
(1) Conferencia internacional sobre armonización de los requisitos técnicos
para el registro de medicamentos de uso humano.
(2) DO L 193 de 17.7.1991, p. 30.
(3) DO L 121 de 1.5.2001, p. 34.
(4) DO L 15 de 17.1.1987, p. 29.
(5) DO L 145 de 11.6.1988, p. 35.
(6) Véase la página 1 del presente Diario Oficial
(7) Véase la página 24 del presente Diario Oficial.
(8) DO L 106 de 17.4.2001, p. 1.
(9) DO L 11 de 14.1.1978, p. 18.
(10) DO L 237 de 10.9.1994, p. 13.
(11) DO L 313 de 13.12.2000, p. 22.
(12) DO L 55 de 11.3.1995, p. 15.
(13) DO L 214 de 24.8.1993, p. 1."
ANEXO II
PARTE A
Directivas derogadas, con sus modificaciones sucesivas (contempladas en el artículo
128)
Directiva 65/65/CEE del Consejo (DO 22 de 9.2.1965, p. 369/65)
Directiva 66/454/CEE del Consejo (DO 144 de 5.8.1966, p. 2658/66)
Directiva 75/319/CEE del Consejo (DO L 147 de 9.6.1975, p. 13)
Directiva 83/570/CEE del Consejo (DO L 332 de 28.11.1983, p. 1)
Directiva 87/21/CEE del Consejo (DO L 15 de 17.1.1987, p. 36)
Directiva 89/341/CEE del Consejo (DO L 142 de 25.5.1989, p. 11)
Directiva 92/27/CEE del Consejo (DO L 113 de 30.4.1992, p. 8)
Directiva 93/39/CEE del Consejo (DO L 214 de 24.8.1993, p. 22)
Directiva 75/318/CEE del Consejo (DO L 147 de 9.6.1975, p. 1)
Directiva 83/570/CEE del Consejo
Directiva 87/19/CEE del Consejo (DO L 15 de 17.1.1987, p. 31)
Directiva 89/341/CEE del Consejo
Directiva 91/507/CEE de la Comisión (DO L 270 de 26.9.1991, p. 32)
Directiva 93/39/CEE del Consejo
Directiva 1999/82/CE de la Comisión (DO L 243 de 15.9.1999, p. 7)
Directiva 1999/83/CE de la Comisión (DO L 243 de 15.9.1999, p. 9)
Directiva 75/319/CEE del Consejo
Directiva 78/420/CEE del Consejo (DO L 123 de 11.5.1978, p. 26)
Directiva 83/570/CEE del Consejo
Directiva 89/341/CEE del Consejo
Directiva 92/27/CEE del Consejo
Directiva 93/39/CEE del Consejo
Directiva 2000/38/CE de la Comisión (DO L 139 de 10.6.2000, p. 28)
Directiva 89/342/CEE del Consejo (DO L 142 de 25.5.1989, p. 14)
Directiva 89/343/CEE del Consejo (DO L 142 de 25.5.1989, p. 16)
Directiva 89/381/CEE del Consejo (DO L 181 de 28 6.1989, p. 44)
Directiva 92/25/CEE del Consejo (DO L 113 de 30.4.1992, p. 1)
Directiva 92/26/CEE del Consejo (DO L 113 de 30.4.1992, p. 5)
Directiva 92/27/CEE del Consejo
Directiva 92/28/CEE del Consejo (DO L 113 de 30.4.1992, p. 13)
Directiva 92/73/CEE del Consejo (DO L 297 de 13.10.1992, p. 8)
PARTE B
Lista de plazos de transposición al Derecho nacional (contemplados en
el artículo 128)
ANEXO III
Incluye un cuadro de correspondencias.