Procedimientos de comunicación en materia de farmacovigilancia de medicamentos de uso humano entre la Industria Farmacéutica y la Agencia Española del Medicamento.
Como consecuencia de la publicación del Estatuto de la Agencia Española del Medicamento (Real Decreto 520/1999, BOE 31/03/1999) se ha reorganizado el personal adscrito a tareas de Farmacovigilancia.
Para todos los efectos, la unidad de la Agencia Española del Medicamento (AEM) responsable de todas las tareas de farmacovigilancia es la División de Farmacoepidemiología y Farmacovigilancia (en adelante Div. FE y FV), adscrita a la Subdirección General de Seguridad de Medicamentos. Esta División se ubica en Majadahonda (Madrid) y le competen todas las tareas de interlocución con la Industria farmacéutica en temas de farmacovigilancia, además de actuar como Centro Coordinador del Sistema Español de Farmacovigilancia (SEFV).
La Agencia Europea para la Evaluación de Medicamentos (EMEA) ha hecho públicas las Guías de Farmacovigilancia en el Notice to Marketing Authorisation Holders para que sirvan como documento de referencia de los Laboratorios farmacéuticos en materia de farmacovigilancia. El texto fue elaborado por el Grupo de Trabajo de Farmacovigilancia Europeo (Pharmacovigilance Working Party), una vez consultada la Industria farmacéutica, y se adoptó por el Comité de Especialidades Farmacéuticas (CPMP) de la EMEA en la reunión de enero de 1999. Este documento se ha remitido a la Comisión Europea para su futura inclusión en el Volume IX of Rules Governing Medicinal Products in the European Union y está disponible en la siguiente dirección de Internet de la EMEA:
http://www.eudra.org/humandocs/PDFs/PHVWP/010899en.pdf
Por todo lo anterior, se ha decidido la publicación de esta Circular que, al incorporar las ultimas recomendaciones europeas en materia de farmacovigilancia, sirve de actualización de lo contemplado en la "Guía para la Industria Farmacéutica en España" de marzo de 1995 (Circular 31/95 de Farmaindustria), documento de referencia hasta este momento y que la presente Circular sustituye.
1 Ambito de aplicación
La presente Circular tiene por objeto clarificar el procedimiento que debe seguirse en las actividades de farmacovigilancia en el seno de la AEM. Los detalles de aplicación de la normativa se encuentran reflejados en el Notice to Marketing Authorisation Holders citado anteriormente que, junto con esta Circular, constituye el documento de referencia para el cumplimiento de las obligaciones en farmacovigilancia por parte de los Laboratorios farmacéuticos en España. Cualquier modificación que se realice en este documento y sea adoptada por el CPMP, debe entenderse que sustituye al texto de aplicación antes citado.
Las obligaciones en farmacovigilancia se aplican a todos los medicamentos autorizados, con independencia de su fecha de autorización.
2. Responsabilidades de la Industria farmacéutica
La responsabilidad legal derivada del cumplimiento de las obligaciones de farmacovigilancia recae siempre en el titular de la autorización de comercialización (TAC), que como tal figure inscrito en el registro de especialidades farmacéuticas. En caso de que haya un acuerdo de comercialización con otra compañía, las funciones de farmacovigilancia las podrá realizar el TAC por su cuenta o a través del Laboratorio comercializador. Este acuerdo se pondrá en conocimiento de la Div de FE y FV.
Los TAC deberán tener designada una persona cualificada como Responsable de Farmacovigilancia (RFV) ubicada en España. Este RFV será el interlocutor válido ante la Autoridad reguladora en materia de farmacovigilancia. Se comunicará el nombre y dirección postal y electrónica (esta última si la hubiere) de la persona designada, así como los posteriores cambios, a la Div de FE y FV de la AEM bien por correo postal, por fax o por correo electrónico (ver dirección de contacto en la sección nº 9) en el plazo de 30 días desde la entrada en vigor de las presentes instrucciones.
Cada RFV asumirá las responsabilidades marcadas por la normativa en materia de farmacovigilancia, tales como: establecimiento de un sistema que asegure el registro y mantenimiento de toda la información sobre sospechas de reacciones adversas (RA) a las especialidades farmacéuticas de las que su compañía es TAC, y cumplimiento de los procedimientos y preparación de informes que precisen las autoridades competentes.
3. Notificación expeditiva de casos individuales de sospechas de reacción adversa (RA) a medicamentos autorizados en España
Se notificarán directamente a la Div. de FE y FV de la AEM en un plazo
máximo de 15 días naturales, a contar desde que la Compañía
tenga conocimiento del caso.
Se contemplan los siguientes situaciones:
a) Las notificaciones de casos de sospechas de reacción adversa (RA) graves sucedidas en España y comunicadas por un Profesional sanitario o que se hayan recogido de la literatura científica mundial, o de cualquier estudio post-autorización- sean de tipo experimental (ensayos clínicos en las condiciones de uso autorizadas) o de tipo observacional- de las que el Laboratorio tenga conocimiento, se notificarán en el formulario de notificación individual (ver Anexo I). Se enviarán preferiblemente por fax a la Div. de FE y FV. Solo se aceptarán notificaciones escritas en castellano. Si se desea enviarlas por correo postal al Registro Central de la AEM (ver dirección de contacto en la sección nº 9), deberán ir acompañadas obligatoriamente de la carátula que se muestra en el anexo II, marcando la casilla correspondiente al tipo de notificación que se envía. Puede utilizarse una única carátula para enviar más de una notificación, y asociadas a medicamentos diferentes, siempre que sean del mismo tipo (graves y ocurridas en España).
Para las sospechas de RA graves recogidas en el transcurso de un estudio post-autorización y atribuibles al medicamento comparador o a la medicación concomitante, se recomienda que el promotor del estudio comunique la citada sospecha a los TAC concernidos, y que éstos las notifiquen de la forma antes señalada.
b) Las notificaciones de casos de sospechas de RA graves y a la vez inesperadas, que ocurran en países de fuera de la Unión Europea, y comunicadas por un profesional sanitario o que se hayan recogido de la literatura científica mundial, o de cualquier estudio del que tenga conocimiento el TAC, se notificarán en el formulario CIOMS-I. La información podrá ir escrita en español o inglés. Se enviarán solo por correo postal al Registro Central de la AEM acompañadas obligatoriamente de la carátula que se muestra en el anexo II, marcando la casilla correspondiente al tipo de notificación que se envía. Puede utilizarse una única carátula para enviar más de una notificación, y asociadas a medicamentos diferentes, siempre que sean del mismo tipo (graves e inesperadas y ocurridas fuera de la Unión Europea).
Cuando la sospecha de RA proceda de la literatura, en los dos casos anteriores, se acompañará al formulario correspondiente una fotocopia de la publicación original (traducida al castellano o al inglés si estuviera en otra lengua) y siempre de forma que se pueda comprobar la referencia bibliográfica (título de la revista, año, volumen y páginas).
4. Informes periódicos de seguridad (IPS)
Deben realizarse informes periódicos de seguridad (IPS) para cada una de las especialidades farmacéuticas autorizadas en España, con independencia del procedimiento de registro utilizado y de la fecha de autorización.
De acuerdo con la normativa europea y para facilitar la elaboración y evaluación de los IPS, cada Laboratorio podrá integrarlos por principio activo, pero en este caso la periodicidad del único IPS resultante vendrá marcada por la fecha de autorización de la especialidad farmacéutica más reciente.
En el caso de que varias compañías deseen presentar un IPS común por principio activo, debido a que entre las mismas se hubieran establecido acuerdos, cada IPS recogerá toda la información de las especialidades farmacéuticas implicadas, estando marcada su periodicidad por la fecha de autorización de la especialidad farmacéutica más reciente. Además, deberá presentarse a la Div de FE y FV un escrito firmado por las compañías afectadas en el que figuren las especialidades para las que se va a presentar un IPS común por principio activo.
Los IPS se presentarán siempre por duplicado en el Registro Central de la AEM e irán dirigidos a la Div de FE y FV. Cada ejemplar se acompañará obligatoriamente de una carátula en castellano (ver modelo de hoja de presentación de IPS en Anexo III) con los pertinentes datos identificativos. Todos los IPS deben siempre adjuntar la ultima ficha técnica (y/o prospecto) autorizada en España, con fotocopia del documento de autorización.
La frecuencia de presentación de IPS es la siguiente:
a)- en cualquier momento a solicitud de las Autoridades sanitarias.
b)- cada 6 meses durante los dos primeros años, a contar desde la fecha
de autorización.
c)- anualmente los dos años siguientes.
d)- en el momento de la solicitud de la primera revalidación.
e)- quinquenalmente junto con las solicitudes de revalidación.
Los IPS incluirán toda la información disponible desde el punto
de cierre de datos del informe anterior y se deberán presentar en un
plazo máximo de 60 días a contar desde el nuevo punto de cierre
de datos. Si el TAC tiene elaborado un IPS que abarca un periodo menor, se aceptará
la presentación del IPS disponible adjuntando un anexo con todos los
datos internacionales recogidos hasta abarcar el periodo correspondiente, junto
con una evaluación de los mismos.
La fecha de autorización que debe considerarse para la elaboración
de los IPS es la siguiente, según el procedimiento de registro empleado
para su autorización:
![]() Procedimiento centralizado:
la fecha de autorización de la Comisión Europea.
Procedimiento centralizado:
la fecha de autorización de la Comisión Europea.
![]() Procedimiento de reconocimiento
mutuo: la fecha de nacimiento europea (sólo para los IPS que deben presentarse
durante los cinco primeros años desde la autorización, ya que
los IPS quinquenales se presentarán junto con la solicitud de revalidación).
En estos casos, se enviarán los IPS que la compañía elabore
tras la finalización del proceso de reconocimiento mutuo, aunque formalmente
el medicamento aún no estuviera comercializado en España.
Procedimiento de reconocimiento
mutuo: la fecha de nacimiento europea (sólo para los IPS que deben presentarse
durante los cinco primeros años desde la autorización, ya que
los IPS quinquenales se presentarán junto con la solicitud de revalidación).
En estos casos, se enviarán los IPS que la compañía elabore
tras la finalización del proceso de reconocimiento mutuo, aunque formalmente
el medicamento aún no estuviera comercializado en España.
![]() Procedimiento nacional: la
fecha de la autorización de comercialización otorgada por la AEM.
Procedimiento nacional: la
fecha de la autorización de comercialización otorgada por la AEM.
Para posibilitar la presentación del IPS correspondiente al quinto año
después de la autorización junto con la solicitud de revalidación
de la especialidad farmacéutica, dicho informe podrá abarcar un
periodo inferior a un año, pero siempre igual o superior a seis meses.
Las especialidades farmacéuticas que se autoricen por procedimientos de reconocimiento mutuo o centralizado seguirán el régimen previsto en los mismos en lo referente a remisión de IPS y revalidaciones quinquenales. Para el resto de especialidades, opcionalmente y al efecto de armonizar fechas de cierre de datos a escala internacional, cabe solicitar un adelanto de la fecha de revalidación, sin que ello suponga en, ningún caso, una modificación de la periodicidad de presentación de los IPS. Esto es, un adelanto de la 1ª revalidación no exime de la obligación de presentar los IPS requeridos durante los cinco primeros años de autorización y, una vez superado este periodo, se irán presentando los IPS de las revalidaciones sucesivas tomando como referencia la nueva fecha de revalidación otorgada. En el caso de que se adelante alguna de las revalidaciones posteriores a la primera, los IPS que se deben presentar abarcarán intervalos de cinco años a contar desde la nueva fecha de revalidación otorgada. La Compañía que opte por hacer uso de esta posibilidad de adelanto de la revalidación, lo hará constar en el apartado de "aclaraciones adicionales" de la hoja de presentación (ver anexo III) del IPS correspondiente.
Cualquier solicitud de presentación de un IPS en periodos o fechas distintos de los establecidos en los primeros 5 años desde su autorización- y siempre teniendo en cuenta las posibles fechas de autorización antes mencionadas -, deberá realizarse en escrito aparte dirigido a la Div de FE y FV que se entregará en el momento de presentar la solicitud de autorización de registro en la AEM o bien, después de otorgado el registro, mediante la solicitud de una Modificación de Tipo II, según la Circular Nº 18/97 de la DGFPS, en su instrucción quinta, letra D, punto 4º, ya que se considera como "modificación de importancia mayor". Dicha modificación será presentada en el Registro Central de la AEM (c/ Huertas 75, Madrid) y dirigida a la Div. de FE y FV. A efecto del pago de tasas, este procedimiento corresponde al Tipo 1.08.990.
Estas solicitudes deberán acompañarse siempre de una justificación pormenorizada de los motivos en los que se fundamente la petición, además de una copia de la Ficha técnica y/o prospecto (autorizada o propuesta).
En el apartado 4.2.5.2 del Notice to Marketing Authorization Holders se recogen algunos supuestos en los que se podría considerar la solicitud de una modificación de la periodicidad o de las fechas de presentación de los IPS durante los primeros cinco años desde la autorización.
En las directrices ICH/E2C, que entraron en vigor en la Unión Europea el 18 de junio de 1997 después de aprobarse por el CPMP el 18 de diciembre de 1996, se introduce el concepto nuevo de RA referenciada / no referenciada. Una RA se considerará referenciada si se describe en la Información Básica de Seguridad del Producto, conocida también como Company Core Safety Information, y que a su vez está contenida en el Company Core Data Sheet. Este último puede ser idéntico al SPC o Ficha Técnica autorizada por las Autoridades reguladoras europeas o españolas.
Los listados (line listings) de RA que se incluirán en los IPS, deben recoger:
a)- las sospechas de RA "graves" y las RA "no graves-no referenciadas" recibidas espontáneamente de profesionales sanitarios o publicadas en la literatura científica mundial.
b)- las sospechas de RA "graves" procedentes de estudios (publicados o no) o de un uso compasivo de la especialidad farmacéutica.
c)- las sospechas de RA "graves" procedentes de autoridades reguladoras, identificadas convenientemente con un asterisco.
Las sospechas de RA "no graves-referenciadas" se comunicarán, siempre que la autoridad reguladora lo requiera, en un listado aparte.
En todos los casos sólo se aceptarán informes escritos en castellano o en inglés.
5. Estudios de Seguridad post-autorización (ESPA) promovidos por
compañías farmacéuticas
Un ESPA es un estudio farmacoepidemiológico o un ensayo clínico
realizado de acuerdo a las condiciones de uso establecidas en la Ficha Técnica,
con el objetivo de identificar o cuantificar un aspecto concreto del perfil
de seguridad de un medicamento autorizado. Se considerará también
un ESPA al estudio en el que el número de pacientes a incluir pueda añadir
información relevante a los datos de seguridad ya existentes del medicamento.
Las disposiciones que siguen son de aplicación a los ESPA promovidos de forma completa o parcial por una Compañía farmacéutica en los que el medicamento es suministrado por el Laboratorio promotor o bien prescrito en la forma habitual, tanto en el ámbito hospitalario como en el de atención primaria.
En línea con lo establecido desde 1990 mediante la Circular nº 18/90 de la DGFPS, y tal como se recoge en las directrices europeas, y siempre para garantizar los objetivos científicos de los ESPA, se establecen los siguientes cauces de relación entre los Laboratorios farmacéuticos y las Autoridades reguladoras:
a)- Se recomienda discutir el protocolo del estudio con las autoridades reguladoras (técnicos de la Div. de FE y FV (AEM)), y expertos independientes, en fases precoces del mismo.
b)- El protocolo del estudio, junto con el material informativo que se envía a los profesionales sanitarios, se presentará en la AEM, dirigido a la Div. de FE y FV, al menos un mes antes del inicio previsto del estudio.
c)- La compañía comunicará la fecha efectiva de comienzo del estudio y enviará un informe de seguimiento cada seis meses, o antes si así se solicita. Asimismo la Compañía informará de forma inmediata sobre cualquier incidencia relevante (interrupción, modificación sustancial del protocolo…) que pueda producirse en el transcurso del estudio. Todas estas comunicaciones se presentarán en la AEM e irán dirigidas a la Div. de FE y FV.
d)- Se cumplirán los requisitos de notificación de RA detectadas durante el estudio y establecidos en secciones precedentes (ver sección 3).
e)- La compañía presentará en la AEM un informe final del estudio entre los tres y seis meses siguientes a su finalización. Dicho informe se dirigirá a la Div. de FE y FV.
f)- Se recomienda que todos los protocolos de los ESPA sean sometidos a la consideración de un Comité Etico de Investigación Clínica, siendo esto obligatorio cuando se solicita información de forma directa al paciente o se realizan pruebas adicionales fuera de la práctica clínica habitual, o bien cuando los tratamientos se asignen de forma sistemática.
6. Evaluación continuada de la relación beneficio/riesgo tras la autorización.
En materia de seguridad de los medicamentos existe una responsabilidad compartida entre las Autoridades reguladoras y los TAC.
En línea con este compromiso, se espera que los TAC pongan en conocimiento de la Subdirección General de Seguridad de Medicamentos (con copia a la Div. de FE y FV) de la AEM sin tardanza, y por escrito, cualquier información relevante que afecte a la seguridad de los medicamentos autorizados (o en tramite de autorización) en España.
Del mismo modo actuará la AEM cuando conozca nueva información relevante que afecte a la seguridad de un medicamento. La AEM podrá solicitar al TAC un informe actualizado de evaluación de la relación beneficio-riesgo, siempre que lo considere oportuno.
Cualquier carta dirigida a los profesionales sanitarios relativa a un problema
de seguridad de un medicamento, deberá acordarse previamente en sus términos
con la Div. de FE y FV.
7. Provisión de información del Sistema Español de Farmacovigilancia
(SEFV) a los Titulares de Autorizacion de comercialización (TAC).
La Div. de FE y FV de la AEM realizará quincenalmente envíos
por correo a cada TAC con la información individualizada de las notificaciones
de sospechas de RA graves incorporadas a la base de datos FEDRA, en las que
sus especialidades farmacéuticas hayan sido consideradas como sospechosas.
Las aclaraciones a los envíos quincenales deberá solicitarse siempre
por escrito.
Toda petición de información sobre RA que no cumplan los criterios
de envío expeditivo (RA graves en 15 días) se hará a través
de una solicitud de Expedición de Certificaciones oficiales en materia
de farmacovigilancia, previo pago de las tasas correspondientes. Una vez pagadas
las tasas se presentará la solicitud en la AEM, dirigida a la Div. de
FE y FV junto con una fotocopia del pago realizado.
8. Modificaciones en la condiciones de autorización de las especialidades farmaceúticas de uso humano, relativas a farmacovigilancia.
En el marco de la redistribución de funciones llevada a cabo en la AEM, la Div. de FE y FV ha asumido la evaluación de las siguientes modificaciones en las condiciones de Autorización (en adelante Variaciones), contempladas en la Circular nº 18/97 de la DGFPS:
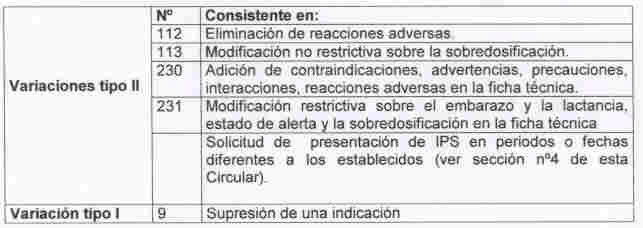
Las correspondientes solicitudes (por duplicado) se presentarán en el
Registro Central de la AEM, dirigidas a la Div de FE y FV y previo pago de las
correspondientes tasas. Con objeto de agilizar su evaluación, se recuerda
la necesidad de adjuntar datos justificativos del cambio que se propone, además
de una propuesta de ficha técnica y prospecto ya modificados, reseñando
en el texto los cambios solicitados.
Cualquier petición de información sobre la evaluación científica una determinada variación se solicitará por correo postal o fax a la Div de FE y FV.
De acuerdo a lo especificado en la sección nº 2 de esta Circular,
la persona de contacto de los TAC con la Div. de FE y FV en lo referente a cuestiones
técnicas sobre las Variaciones arriba mencionadas es el Responsable de
Farmacovigilancia.
9. Dirección de contacto
Toda la información que se cita en esta Circular se enviará normalmente por correo postal, salvo que se indique otra forma. Para casos excepcionales, de comunicación urgente de un riesgo importante, se utilizará el fax, el teléfono y/o el correo electrónico de la Secretaría de la División de FE y FV:
Direccción Postal:
Secretaría de la División de Farmacoepidemiología y Farmacovigilancia.
Agencia Española del Medicamento.
Ctra Majadahonda-Pozuelo km. 2
28220-Majadahonda, Madrid.
Telf: 91. 596 77 11
Fax: 91. 596 78 91
E-mail: fvigilancia@agemed.es
Registro Central de la AEM:
C/ Huertas nº 75
28001- Madrid.
Horario: 8:30 a 14:00 horas, lunes a viernes.
10.Tabla-resumen de la entrada de documentos procedentes de la industria farmacéutica dirigidos a la Div. de FE y FV.

Abreviaturas:
AEM: Agencia Española del Medicamento; Registro Central (ver seccion
nº 9).
Div. FE y FV: División de Farmacoepidemiología y Farmacovigilancia
(ver sección nº 9).
FEDRA: Farmacovigilancia Española, Datos de RA; acrónimo de la
base de datos del SEFV.
IPS: Informe periódico de seguridad.
RA: Reacción Adversa a medicamentos.
SEFV: Sistema Español de Farmacovigilancia.
TAC: Titular de Autorización de Comercialización.
UE: Unión Europea
EL DIRECTOR DE LA AGENCIA ESPAÑOLA DEL MEDICAMENTO
Josep Torrent i Farnell
ANEXO I
FORMULARIO DE NOTIFICACION INDIVIDUAL



